2014年度 京都工芸繊維大学 遺伝子資源学実習および演習 バイオインフォマティクス基礎編 『高速シーケンサーの大規模データをもちいる遺伝子発現解析実習および演習』 担当者 北島 佐紀人 (京都工芸繊維大学 応用生物学部門) ver. 2014.09.11 |
初めに
この文書は、京都工芸繊維大学で開催される大学院講義”遺伝子資源学実習および演習”(開催日 2014年9月10日~12日)の一部で使用するテキストである。このテキストがカバーする実習では、次世代シーケンサーあるいは高速シーケンサーと呼ばれる装置から出力される膨大な数の塩基配列をとりあつかうためのバイオインフォマティクス的スキルの取得を目指す。モデルケースとして取り上げたのはmRNA-seqと呼ばれる解析手法で、この方法ではどのような遺伝子が発現しているかを網羅的にしらべる。バイオ解析ツールの使い方だけでなく、Linuxのコマンドとシェルスクリプトの初歩についても学べると思う。
この実習の担当教員でテキストの筆者である北島は、バイオインフォマティクスあるいはコンピュータプログラミングの専門家ではない。ここに書いたことを筆者自身が学ぶ過程では、各ツールの説明書だけでなく、インターネット上で見つけた多くの研究者と大学院生の講習テキストあるいはブログが役立った。簡単な内容だが、筆者と同じように勉強中の人々にとって少しでも参考にもなればと思い、このテキストを当分の間インターネットで閲覧できるようにしておく。
テキスト中の間違いなどに関して、もし情報をご提供いただける方がいらっしゃったら、以下までご連絡ください。
北島佐紀人
Email; sakitoあとまーくkit.ac.jp(あとまーくは@)
研究室ブログ;http://ameblo.jp/shokubutsu-bunshi-kit/
1. Bio-Linux 8と解析ツールの準備 |
(1-1) Bio-Linux 8の準備
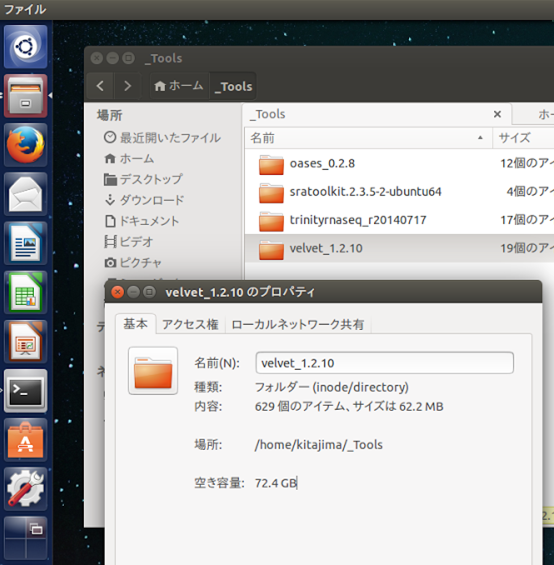
Bio-Linux 8は、Linuxの一種”Ubuntu 14.04 LTS”をベースにして、これに各種バイオ解析ツール群を組み込んだものだ。無償で入手できるので、だれでも手元のPCにインストールして自由にバイオ解析をできる。インストールするには、Bio-Linux 8のサイト( http://environmentalomics.org/bio-linux/ )からBio-Linux 8のISOファイルをダウンロードして、imgBurnというソフトでDVDに焼いた後、PCをそのDVDから起動すれば、後はボタンを押していくだけよい。使用言語に日本語を選べば、日本語を入力できるようになる。バイオ解析ツール以外の各種アプリの使用方法あるいはOSの設定方法は、ubuntuに関する情報が役に立つ。ウィンドウシステムの操作方法は、Windowsとよく似ているので、すぐ分かると思う。画面左にアイコンが並んでいるが、これはWindowsのタスクバーに相当するもので、Ubuntuではランチャーと呼んでいる。ファイルマネージャと、この実習で使うアプリのいくつかは、すでにここに登録されている。ここに登録されていないアプリの起動方法は独特なので、ここで説明しておく。ランチャーの一番上に見えているアイコン(”コンピューターとオンラインリソースを検索”)をクリックすると検索窓が現れるので、ここに、キーワードを入力する。例えば”エディタ”と入力すると、図のように”テキストエディター”というアイコンが現れる。これをクリックすれば起動する。ランチャーに登録しておくことも可能だ。
ブラウザで以下のリンク先を見ると、Bio-Linux 8にはどんなバイオ解析ツールがインストールしてあるのか分かる。
http://environmentalomics.org/bio-linux-software-list/
本実習では、これらに加えてsratoolkit、Trinity、velvetおよびoasesというツールを使用する。解析に先立って、まずはこれらを自分でインストールしてみよう。
(1-2) sratoolkitのインストール
ブラウザで http://www.ncbi.nlm.nih.gov/Traces/sra/?view=software にアクセスする。ホームページを見るとNCBI SRA Toolkitの“Ubuntu Linux 64 bit architecture”というリンクがあるので、これをクリックすると”ダウンロード”という名前のフォルダに圧縮ファイルがダウンロードされる。これを選んで右クリックすると、”ここに展開する”という項目が現れるので、それを選ぶ。 展開されて出来たディレクトリ"sratoolkit.2.3.5-2-ubuntu64”は、すきな場所に置いて差し支えないが、今回は、ホームディレクトリに"_Tools”というディレクトリを新規に作成して、その中に置いておく。ついでにディレクトリ名を”sratoolkit”と変更しておく。方法は、Windowsの場合と似ているので、説明は必要ないと思う。
(1-3) Trinityのインストール
Trinityのホームページ(http://trinityrnaseq.sourceforge.net/)にアクセスする。ホームページを少し下にスクロールするとDownloadingと書いたリンクがあるので、これをクリックして、最新版(このテキスト執筆時点で”trinityrnaseq_r20140717.tar.gz”という名前のファイル)をダウンロードする。”ダウンロード”ディレクトリに圧縮ファイルがダウンロードされる。上と同様に展開し、出来た"trinityrnaseq_r20140717”ディレクトリを"_Tools”ディレクトリの中に置き、ついでにディレクトリ名を”trinityrnaseq”と変更する。ここから先は、Linuxに不慣れなひとには少々ややこしい。まず、端末という名前のアプリを起動して、cdコマンドでTrinityのディレクトリに移動する。以下に示す青色の部分が自分でタイプするところだ。いちいち手打ちしなくても、このテキストからコピペすればよい。ただし、重大な注意点がある。通常のペーストのショートカットキーはWindowsの場合と同じcontrol + pだが、端末の中に限ってはconrol + shift + pだ。ついでに書いておくと、端末に表示されている文字列をコピーする場合は、control + shift + cとタイプする。間違ってconrol + cとすると、実行中の処理が強制的に終了する。間違えそうで心配なら、マウスの右クリックメニューを使えばよい。
kitajima@kitajima[kitajima] cd _Tools/trinityrnaseq |
入力したらエンターキーをおす。すると、[ ]の中が変わってtrinityrnaseqとなったはずだ。
kitajima@kitajima[trinityrnaseq] |
これで、カレントディレクトリが、ホームディレクトリ(最初にいたところ)の下の、_Toolsというディレクトリの下の、trinityrnaseqになった。試しに、”ls” (エルエス)とタイプしてみると、このディレクトリ内のファイル一覧を表示する。
kitajima@kitajima[trinityrnaseq] ls Analysis docs Inchworm notes Release.Notes trinity-plugins Butterfly galaxy-plugin LICENSE.txt PerlLib sample_data util Chrysalis htc_conf Makefile README Trinity |
ファイルマネージャでも同じディレクトリの中身を観ることができるので、たしかにこのディレクトリを表示させていることが確認できる。次に、”make”とタイプすると、このディレクトリのMakefileという名前のファイルの内容に従ってインストールが始まる。待つこと数分。
kitajima@kitajima[trinityrnaseq] make Using gnu compiler for Inchworm and Chrysalis cd Inchworm && (test -e configure || autoreconf) \ && ./configure --prefix=`pwd` && make install (中略) JellyFish: has been Installed Properly Inchworm: has been Installed Properly Chrysalis: has been Installed Properly QuantifyGraph: has been Installed Properly GraphFromFasta: has been Installed Properly ReadsToTranscripts: has been Installed Properly fastool: has been Installed Properly parafly: has been Installed Properly slclust: has been Installed Properly collectl: has been Installed Properly kitajima@kitajima-BioLinux[trinityrnaseq] |
と表示され、元の入力待ち状態になれば完了。試しに、”perl Trinity”とタイプしてみると、下のように表示されるはずだ。
kitajima@kitajima[trinityrnaseq] perl Trinity ############################################################################### # # ______ ____ ____ ____ ____ ______ __ __ # | || \ | || \ | || || | | # | || D ) | | | _ | | | | || | | # |_| |_|| / | | | | | | | |_| |_|| ~ | # | | | \ | | | | | | | | | |___, | # | | | . \ | | | | | | | | | | | # |__| |__|\_||____||__|__||____| |__| |____/ (以下略) |
ファイル”Trinity”は、アプリケーションではなく、perlというプログラミング言語のスクリプトなので、このようにタイプした(このファイルをテキストエディタで開いて、1行めをみるとperlスクリプトであることが分かる。)。最後に、次にようにタイプしてスタックサイズの制限を解除する。これを忘れると計算中にエラーすることがあるそうだ。
kitajima@kitajima[trinityrnaseq] ulimit -s unlimited |
余談だが、 ”cd ~”とタイプすると、カレントディレクトリがホームディレクトリに移る。ここで、同じように”perl Trinity”とタイプするとエラーする。これは、カレントディレクトリであるホームディレクトリにTrinityというファイルがないためだ。
kitajima@kitajima[trinityrnaseq] cd ~ kitajima@kitajima[kitajima] perl Trinity Can't open perl script "Trinity": そのようなファイルやディレクトリはありません kitajima@kitajima[kitajima] |
しかし、ファイルの置いてあるディレクトリまで含めて指定するとうまく動く。
kitajima@kitajima[kitajima] perl ~/_Tools/trinityrnaseq/Trinity ############################################################################### # # ______ ____ ____ ____ ____ ______ __ __ # | || \ | || \ | || || | | # | || D ) | | | _ | | | | || | | # |_| |_|| / | | | | | | | |_| |_|| ~ | # | | | \ | | | | | | | | | |___, | # | | | . \ | | | | | | | | | | | # |__| |__|\_||____||__|__||____| |__| |____/ (以下省略) |
“~”とは、ホームディレクトリを意味する記号だ。したがって、”perl ~/_Tools/trinityrnaseq/Trinity”は、perlはホームディレクトリの下の_Toolsディレクトリの下のtrinityrnaseqディレクトリの下のTrinityというファイルを実行せよ、という意味になる。このように目的のファイルまでの道筋(”パス”と呼ぶ)を毎回指定するのは面倒なので、あらかじめOSにホームディレクトリからのパスを登録しておくことも可能だが、それも面倒なので今回は省略する。方法を知りたいひとは、”Linux, パスを通す方法”というようなキーワードでググってみるとよい。なお、このファイルがperl等により実行可能なスクリプトである、とOS側に認識されている場合は、冒頭の”perl “を省略しても動作する。
(1-4) velvetとOasesのインストール
velvetとOasesはすでにBio-Linux 8にインストールされているので、これらを使ってもよい。しかし、k-merというオプションの値の上限が31に設定されている。k-merの値は、実際にはもっと大きな値に設定して使うことになるので、上限値を上げるためにインストールから自分でやることにする。velvetとOasesはそれぞれ以下のホームページからダウンロードする。
https://www.ebi.ac.uk/~zerbino/velvet/
http://www.ebi.ac.uk/~zerbino/oases/
Current version : xxxxというようなリンクがあるので、これをクリックすると、ダウンロードディレクトリに圧縮ファイルがダウンロードされる。このテキスト執筆時点で、それぞれのバージョンは1.2.10および0.2.08である。上と同様の方法で展開したあと、ホームディレクトリの_Toolsディレクトリに移し、ディレクトリ名をそれぞれ”velvet”と”oases”と変更する。まず、velvetをインストールする。以下のように、カレントディレクトリをvelvetのディレクトリに変更して、makeする。makeのあとにMAXKMERLENGTH=99と付け加えるのは、k-merの上限を99に設定するためだ。
kitajima@kitajima[velvet] cd ~/_Tools/velvet kitajima@kitajima[velvet] make MAXKMERLENGTH=99 |
以下のように表示されれば成功である。試しに”velveth -help”とでも入力すると実行されたのが確認できるはずだ。
kitajima@kitajima[velvet] make MAXKMERLENGTH=99 rm obj/*.o obj/dbg/*.o rm: `obj/*.o' を削除できません: そのようなファイルやディレクトリはありません rm: `obj/dbg/*.o' を削除できません: そのようなファイルやディレクトリはありません (中略) gcc -Wall -m64 -O3 -o velvetg obj/tightString.o obj/graph.o obj/run2.o obj/fibHeap.o obj/fib.o obj/concatenatedGraph.o obj/passageMarker.o obj/graphStats.o obj/correctedGraph.o obj/dfib.o obj/dfibHeap.o obj/recycleBin.o obj/readSet.o obj/binarySequences.o obj/shortReadPairs.o obj/scaffold.o obj/locallyCorrectedGraph.o obj/graphReConstruction.o obj/roadMap.o obj/preGraph.o obj/preGraphConstruction.o obj/concatenatedPreGraph.o obj/readCoherentGraph.o obj/utility.o obj/kmer.o obj/kmerOccurenceTable.o obj/allocArray.o obj/autoOpen.o -lz -lm kitajima@kitajima[velvet] |
次に、oasesをインストールする。カレントディレクトリをoasesに変更し、以下のようにしてmakeする。
kitajima@kitajima[kitajima] cd ~/_Tools/oases kitajima@kitajima[oases] make 'VELVET_DIR=/home/kitajima/_Tools/velvet' 'MAXKMERLENGTH=99' |
'VELVET_DIR=/home/kitajima/_Tools/velvet'はvelvetの置き場所を示している。/home/kitajimaの部分は”~”と同じ意味なので、~/_Tools/velvetと書いても良さそうに思えるが、oasesのマニュアルに、~はダメ、と書いてある。kitajimaの部分は、ユーザーごとに違うので書き換えないといけない。ファイルマネジャーでvelvetのディレクトリを選び、右クリックで”プロパティ”を選ぶと、ユーザーそれぞれの場合のパスが表示されるので、それをコピペすればよい。'MAXKMERLENGTH=99'は、k-merの上限値を99にするよう指定している。
以下のように表示されれば成功である。
(前略) Output written on OasesManual.pdf (10 pages, 116747 bytes). Transcript written on OasesManual.log. mv *.pdf ../../ make[2]: ディレクトリ `/home/kitajima/_Tools/oases/doc/manual' から出ます make[1]: ディレクトリ `/home/kitajima/_Tools/oases/doc' から出ます kitajima@kitajima[oases] |
試しに”~/_Tools/oases/oases --help”とでも入力すると実行されたのが確認できるはずだ。
kitajima@kitajima[oases] ~/_Tools/oases/oases --help Usage: ./oases directory [options] (中略) -degree_cutoff <integer> : Maximum allowed degree on either end of a contigg to consider it 'unique' (default: 3) Output: directory/transcripts.fa directory/contig-ordering.txt kitajima@kitajima[oases] |
Trinityのところでも述べたように、それぞれのツールのディレクトリがカレントディレクトリである場合をのぞけば、実行ファイルの置き場所をディレクトリから示してあげないと実行できない。例えば、velvethを実行するなら、”~/_Tools/velvet/velveth”とタイプする。(しかし、カレントディレクトリがoasesの時に単純にoases --helpとタイプすると、そんなコマンド知らないよ、みたいなエラーが出た。理由はしらない。)
(1-5) ついでに、OSと各種アプリのアップデートもしておこう。本来、Bio-Linux 8をインストールしたら最初にやるべきことだが、順番が逆になった。端末に順に、”sudo apt-get update”、”sudo apt-get upgrade”とタイプすればよい。パスワードを求められたらログインパスワードを入力する。完了まで少々時間がかかる。
2. 非モデル生物のmRNA-seq解析 |
(2-1) データの準備
高速シーケンサーを使うと、ある生物サンプルから得たmRNAプールの逆転写物を適当なサイズに断片化して、それらを同時に網羅的にシーケンシングすることができる。illumina社のいわゆるショートリードの高速シーケンサーの場合、得られる1本1本の配列データ(“リード”と呼ぶ)は50~百数十塩基長と短い(しかし新機種がリリースされるごとにだんだん長くなっている)が、それが数百万あるいは数千万本も得られる。この配列データを用いて発現解析をする原理は、おおまかには次のようなものだ。それぞれのリードをゲノム配列あるいはmRNAデータベースと比較して、各リードがどの遺伝子あるいはmRNAに一致するか = 由来するか、を調べる。多数のリードが一致した遺伝子あるいはmRNAは発現レベルが高く、逆にほとんどリードが見つからない場合は発現レベルが低いと考えられる。このような解析をmRNA-seqと呼んでいる。
しかし、非モデル生物のような研究事例が少ない生物の場合には、取得したリードを比較すべき配列データベースがないかもしれない。この場合には、取得したリード自身をもちいてmRNAデータベースを作成し、それと各リードを比較する、という方法を取らざるを得ない。本実習の趣旨は、”遺伝資源の研究や利用に必要とされる知識と技術を修得する”ことなので、多様な生物を扱うことを想定して、まずは非モデル生物のケースを学んでみる。
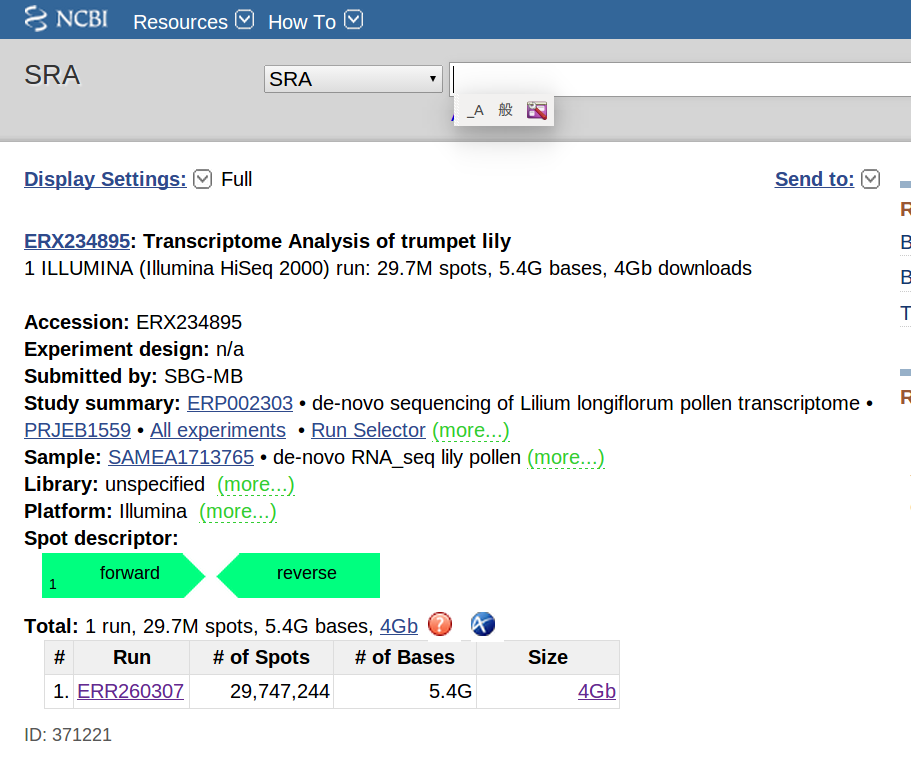
例として、テッポウユリ(Lilium longisporum)のmRNA-seqを行う。NCBIのSRAデータベースに登録されているアクセッションNo. ERX234895 (http://www.ncbi.nlm.nih.gov/sra/ERX234895 )にアクセスする。説明文によると、テッポウユリの花粉よりmRNAを抽出してcDNAとし、それらを約300bpの長さに断片化し、Illumina社の高速シーケンサーHiSeq 2000を用いて、paired endで配列を読んだ、とのことである。Paired endとは、1本のDNA断片の両端から読む解析手法である。このシーケンサーの場合は、それぞれの端から100塩基ずつ読むことができる。
テッポウユリのゲノム配列は登録されていないので、ERX234895に含まれている100塩基長のリードが何の遺伝子に由来するのかわからない。そこで、まずはこれら100塩基長のリード群を互いに比較して、配列の一致する部分を頼りにつなぎあわせていくことにする。この作業を”de novoアッセンブリー”と呼び、出来上がったそれぞれの配列を”コンティグ”と呼ぶ。信頼性の問題は大いにあるものの、作製されたコンティグ = 推定mRNA配列と考える。今回は省略するが、これら推定mRNA配列をBLAST検索すれば、どのようなタンパク質をコードしているか、さらにはその機能も推定できる。次に、こうして出来上がった推定mRNA配列のそれぞれに対して、配列の一致するリードを張り付けていく。この作業を"マッピング”、リードを貼り付けられる配列(この場合は推定mRNA)を”リファレンス配列”と呼ぶ。張り付いたリードの数が多い推定mRNAは、発現量が多い、ということになる。
では、実際にやってみよう。ホームページ左下の表のSizeというカラムにファイルサイズが書いてある。これをクリックすると、ダウンロードページに移る。ファイル名をクリックすると、ダウンロードディレクトリに”ERR260307.sra”という名前のファイルが作成されるはずだ。とりあえず、このファイルを好きなディレクトリに移す。今回は、大学のコンピューターシステムの事情により、~/work/<ユーザー名>に移す。例えば、筆者の場合はアカウント名sakitoなので、~/work/sakitoだ。このテキストでは、これ以降ディレクトリ名あるいはコマンドの中にsakitoという文字が何度か出てくるが、この部分を各自のアカウント名に置き換えてタイプしなければならない。
(2-2) fastq形式への変換
sraファイルのままでは、解析ツールに読み込ませることはできない。そこで、sratoolkitに含まれているfastq-dumpというツールを使ってfastq形式のファイルに変換する。fastq-dumpは、sratoolkitディレクトリのbinというディレクトリの中にあるはずなので、確認してほしい。fastqに変換するには次のようにタイプすればよい。-O以下は、出力先のディレクトリを指定している。コマンド中のsakitoと書いてあるところは、自分のアカウント名に差し替える。
(改行しているように見えるかも知れないが、右端で折り返しただけだ。Linuxのひとつづきのコマンドの途中に改行が入ることはない。)
kitajima@kitajima[kitajima] ~/_Tools/sratoolkit/bin/fastq-dump ~/work/sakito/ERR260307.sra --split-files -O ~/work/sakito |
末尾に付け加えた”--split-files”は、paired endのデータの場合に、ペア同士を別々のファイルに分けるよう指示している。フリーズしたのかと心配になりそうな状態になるが、しばらく待つと次のように表示される。
kitajima@kitajima[kitajima] ~/_Tools/sratoolkit/bin/fastq-dump ~/work/sakito/ERR260307.sra --split-files -O ~/work/sakito Read 29747244 spots for /home/manager/work/sakito/ERR260307.sra Written 29747244 spots for /home/manager/work/sakito/ERR260307.sra kitajima@kitajima[kitajima] |
ファイルマネージャでホームディレクトリをみると、ERR260307_1.fastqおよびERR260307_2.fastqという2つのファイルができているはずだ。それぞれがPaired endの右から読んだデータと左から読んだデータである(右とか左とか言うのは変だが、、、)。試しにERR260307_1.fastqの中身を見てみよう。ファイルサイズが9GBもあるので、ワープロソフトやテキストエディタで開くのは止めた方がよい。代わりに、headコマンドで冒頭の20行だけ表示させてみる。”head -20 ファイル名”とタイプする。
kitajima@kitajima[kitajima] head -20 ~/work/sakito/ERR260307_1.fastq @ERR260307.1 FCD1CNUACXX:8:1101:1465:2041 length=90 NCGCACGGCCCTTGTGCCGGCGACGCATCATTCAAATATCTGCCCTATCAACTTTCGATGGTAGGATAGGGGCCTACCATGGTATTGACG +ERR260307.1 FCD1CNUACXX:8:1101:1465:2041 length=90 #1:BDDFFGHHGFJHIHIIIGIGIIAHIGHGHHHFD>CDFFBDCBC;ACDDDDDDDD?B@=@CC@CC:9??39;?3<ACDDC4:>>::@< @ERR260307.2 FCD1CNUACXX:8:1101:1437:2070 length=90 NCATCTCCGGCTATCTCACATTCACCACCAACTCCAGTGAGCTCACCTCCGCCATCAGTAGTTCAATCATCTCCACCACAAAAGCATCAA +ERR260307.2 FCD1CNUACXX:8:1101:1437:2070 length=90 #1=DDFFFHHHHHJJJJIJJJJGIJJJJIJJJJIJJJGHIJJJJJIJGJGJFGIJJJIJGJHFHGHHCDFFFFFCCEDDDB?AA?CDDDC @ERR260307.3 FCD1CNUACXX:8:1101:1415:2076 length=90 NGGGCAGGGATGGTGAGGGCGCAGAGGAGGCGGCCGGGCTCGACGGTGTCGACGCGGAGGCCTTGGAGGACGAAAGCTTGGTGGAAGTTG +ERR260307.3 FCD1CNUACXX:8:1101:1415:2076 length=90 #1=DDDDFH?DHHCGDGHJ:GHHIBHICHIEHGEEDDD6=??8007&25><(0:8BB&5@<((+4>3?B##################### @ERR260307.4 FCD1CNUACXX:8:1101:1279:2158 length=90 NGCGGCGCGAACTTCTTGCCGGAGCACTCCTTGAGCATGGCCTCGAAGGACCGGGTGACGAACCCGCCAGCTGCGCCCGCCATGACCAGA +ERR260307.4 FCD1CNUACXX:8:1101:1279:2158 length=90 #1=DDDFFHHHHHIGIJIGIIIDHEAHIJJJGIGEHCEEDDFFFDACDDDDDDBB57<:<25@BDD5>;@BCA9<00<>B@-8@(43:AA @ERR260307.5 FCD1CNUACXX:8:1101:1318:2180 length=90 AATGCATTGACCAGGAAGAAGAGACGGCCATGGAGTCCGAGACCACCCGCCGCCAGCTCGCCGGAAGTATCAGGTACATCAGCTACGCCG +ERR260307.5 FCD1CNUACXX:8:1101:1318:2180 length=90 CCCFFFFFHHHHHJJHIIJJIHGJJJJIJJJJJHIJJJJBIIJDIJJJIGFFFDDDDDDDDDBDB@>:@CADDC:>CCDCCC::>:>@B< kitajima@kitajima[kitajima] |
少々見にくいが、fastq形式のファイルでは4行で1つのリードのデータになっている。空白行はない。
@ERR260307.1 FCD1CNUACXX:8:1101:1465:2041 length=90 NCGCACGGCCCTTGTGCCGGCGACGCATCATTCAAATATCTGCCCTATCAACTTTCGATGGTAGG (以下略) +ERR260307.1 FCD1CNUACXX:8:1101:1465:2041 length=90 #1:BDDFFGHHGFJHIHIIIGIGIIAHIGHGHHHFD>CDFFBDCBC;ACDDDDDDDD?B@=@CC@ (以下略) |
1行めは行頭が“@”、3行めは行頭が”+”で、どちらもリードの名称が続く。2行めは塩基配列で、4行目では各塩基の確からしさ(クオリティと呼ぶ)を記号で表している。4行で1つのリードなので、このファイルの行数を数えて4で割ればリード数が分かる。”wc -l ファイル名”とタイプしてみる。wcはワードカウントのコマンドで、”-l”(エル)は行数を表示させるオプションである。
kitajima@kitajima[kitajima] wc -l ~/work/sakito/ERR260307_1.fastq 118988976 ERR260307_1.fastq kitajima@kitajima[kitajima] |
結果は118988976とのことなので、4で割ると29747244、つまりこのファイルには、29747244本のリードがある。paired endなので、もう一方のファイルERR260307_2.fastqにも同じ数のリードが含まれる。
シーケンサーが出力した生のデータの場合、リード配列にはアダプタ配列が含まれているが、公開データの場合はすでに除去済みだと思う。もし、自分でシーケンスして取得した生データの場合は、アダプタ配列を自分で除去しなければならない。
このような巨大データを使うと、この実習時間中に計算が終了しないので、今回は、最初の30万本のリードだけを取り出して使うことにする。最初の30万本ということは、fastqファイルなら最初の120万行ということになる。上でも使ったheadコマンドでERR260307_1.fastqの冒頭の1200000行を取り出し、モニタに表示する代わりに、ERR260307_short_1.fastqというファイルに書き込む。
kitajima@kitajima[kitajima] head -1200000 ~/work/sakito/ERR260307_1.fastq > ~/work/sakito/ERR260307_short_1.fastq kitajima@kitajima[kitajima] wc -l ~/work/sakito/ERR260307_short_1.fastq 1200000 ERR260307_short_1.fastq kitajima@kitajima[kitajima] |
“>”という記号は、この記号の前のコマンドの出力を次のファイルに書き込め、という意味である。wc -lで調べるとERR260307_short_1.fastqの行数は確かに1200000行である。ERR260307_2.fastqについても、同様に冒頭の1200000行を取り出してERR260307_short_2.fastqに出力する。
kitajima@kitajima[kitajima] head -1200000 ~/work/sakito/ERR260307_2.fastq > ~/work/sakito/ERR260307_short_2.fastq kitajima@kitajima[kitajima] |
(2-3) de novoアセンブリ
de novoアセンブリを行うツールは数種類ある。日本語での解説ブログもあるのでググってみるとよい。illuminaシーケンサーが出力する短いリードのアセンブリに筆者が使うのはvelvetとtrinityだが、一長一短だと感じている。Velvetは計算速度が速いが、スプライシングバリアントと称してよく似た配列を不自然なほどたくさん出力する。数うてば当たる、みたいな状態なのか、プロテオミクス解析のタンパク質データベースとして役に立っている。Trinityは、velvetに比べるとスプライシングバリアントらしきものをあまり多く出力しないので出力結果が比較的すっきりしている。しかし、計算パワーを要求するので、低スペックのPCでTrinityを使うのは実用的でないかもしれない。Velvet、Trinityとも、DDBJのサーバーで計算する無料サービスがある( https://p.ddbj.nig.ac.jp/pipeline/Login.do )ので、これを使うのもよい手だ。
参考までに述べると、サンガー法あるいはRoche社の454高速シーケンサーで得られた数100塩基の長い塩基配列を一緒に混ぜてアセンブルしたい場合もあるかもしれない(いわゆるhybrid assemblyあるいはmixed assembly)。その場合は、Bio-Linux 8に標準で組み込まれていないが、MIRA4、あるいはRoche社が無料公開(2014.08.05時点)しているGS de novo assembler (中身はnewblerというツール)というアセンブラが利用できる。ただし、長い配列向けの計算アルゴリズムを用いているため、Trinity以上に計算パワーを要求する。自分のPCで実行すると1ヶ月待っても計算が終わらないかもしれない。Velvetも見かけ上はhybrid assemblyのようなことをでき、しかも計算スピードも速いが、計算に使用しているde Bruijn graphというアルゴリズムがなんちゃららで厳密には長い配列をアセンブルしているとは言えないという話もあるようだ。アルゴリズムについては、以下のブログを見ると良い。
http://shortreadbrothers.blogspot.jp/2011/07/de-novo-transcriptome-454.html
(2-3-1) Trinityによるde novoアセンブリ
Trinityは、実際にはJellyfish、Inchworm、Chrysalis、Butterflyという4つのプログラムを順に実行する。内部でbowtieというプログラムも使用している。各自のデータ形式とPC環境に応じていくつかのオプションを指定する必要がある。詳しくは説明書を読むとよいが、とりあえず以下のものがよくいじるオプションだ。
--seqType; 入力ファイル形式。今回はfastq形式なので、fqと指定する。
--JM; Jellyfishプログラムがk-merという変数の計算時に確保するメモリ領域。大きくし過ぎるとメモリ上のデータがハードディスクに頻繁に退避するので計算速度が低下する。自分のPCに搭載されているメモリ量に応じて変える必要がある。
--SS_lib_type; 今回はPaired endのデータを使用するので、FRと指定する。
--leftおよび--rightで、対になる2つのfastqファイルをそれぞれ指定する。
--CPU; 計算に割り当てるCPUのスレッド数。
--group_pairs_distance; Paired endのリードペアの間の距離の最大値を指定する。今回使用するデータはcDNA断片のサイズが300bp, 両端から100塩基ずつ読んだとのことなので、距離は100塩基と言うことになる。最大値として300を指定しておく。
--bflyHeapSpaceMax; butterflyプログラムに割り当てるJava max heap spaceなる変数で、大きくし過ぎるとメモリ上のデータがハードディスクに頻繁に退避するので計算速度が低下する。自分のPCに搭載されているメモリ量に応じて変える必要がある。
その他必須ではないが、--min_glue, min_iso_ratio, --path_reinforcement_distanceなどを指定するとアッセンブリの厳しさが変わるようなので試してみるとよい。
--output; 結果を出力するディレクトリ。今回は、ホームディレクトリの下のTrinity_sampleディレクトリに出力するよう指定している。繰り返し実行する場合、前回の実行時と同じディレクトリを指定すると誤作動するので、毎回ディレクトリ名を変更する必要がある。
今回は、以下のような条件で実行してみる。
kitajima@kitajima[kitajima] ~/_Tools/trinityrnaseq/Trinity -CPU 3 --seqType fq --JM 3G --bflyHeapSpaceMax 3G --left ~/work/sakito/ERR260307_short_1.fastq --right ~/work/sakito/ERR260307_short_2.fastq --output ~/work/sakito/Trinity_sample --SS_lib_type FR --group_pairs_distance 300 |
すると以下のように表示されて、長い待ち時間となる。
Current settings: time(seconds) unlimited file(blocks) unlimited data(kbytes) unlimited stack(kbytes) 8192 coredump(blocks) 0 memory(kbytes) unlimited locked memory(kbytes) 64 process 514814 nofiles 1024 vmemory(kbytes) unlimited locks unlimited (以下省略) |
もし計算を強制終了したければ、controlキーを押しながらcとタイプする。ちなみに、システムモニターというアプリを起動して、”リソース”タブをクリックするとCPUとメモリの使用状況がリアルタイムに表示される。これを見ると鋭意計算中かどうか、CPUとメモリのどちらが過負荷状態なのか判断できる。最後に以下のように表示されれば、無事に完了。
All commands completed successfully. :-) CMD finished (659 seconds) Tuesday, August 5, 2014: 12:27:38 CMD: /home/kitajima/_Tools/trinityrnaseq/util/support_scripts/print_butterfly_assemblies.pl /home/kitajima/Trinity_sample/chrysalis/component_base_listing.txt > Trinity.fasta.tmp CMD finished (0 seconds) ################################################################### Butterfly assemblies are written to /home/kitajima/Trinity_sample/Trinity.fasta ################################################################### kitajima@kitajima[kitajima] |
ファイルマネージャで見ると、実行時に指定した”Trinity_sample”ディレクトリの中に、”Trinity.fasta”というファイルができているのが分かる。headコマンドで、最初の20行を表示させてみよう。
kitajima@kitajima[kitajima] head -20 ~/work/sakito/Trinity_sample/Trinity.fasta >c0_g1_i1 len=244 path=[26:0-78 104:79-243] CTGCGAGCCTTTTTGTAATGCCTTTGCAACTTGGTGCCGCACCGAGTGTTGCCTCATTCA CATTGATCGAGCAAGATTGCTGTCCAATGCAAGCCTTCTTAACAACTGAAAGGGCATCGT TAGACTCACAACTACCCTTTTTGAATGATTGGCATGCTCCTTGTGGATCTCCGAAACTAG CGAACTCGATATCAGAAATTGACTGGCCACCTTGGCACGATAAAGTAACTTTATTCCCCT CCAG >c1_g1_i1 len=520 path=[27:0-519] CCAAAGCAGAACGAGAAGATCCCGGCCAGGAGCGCATCCACAGCAAGGTGAATCATCTCG AACTTTCTGACACAGAACTCTTGATTGCCAAAGCATACCCTGGAGAACCAATCAAGCAGA (中略) AGCATGCAGAACTGATCGAAGAGCATGGTAGCAGCAGGAG >c3_g1_i1 len=268 path=[27:0-267] CGCCATCAAGTCCCTCGAAAGCGAAACACCGATCTTCCCATCAACATCCTCCTCCTGAGT AACACACCTAAAGCAGTTATCATCAGCACCCTTATGGGTCCTGACAGTATGCACGAGCTG GTACTTAGACCGCCGGCGATCGGAACGTTTATTCGATAAGAGAATTGCAGCTCCACCCAT kitajima@kitajima[kitajima] |
“>”の行が配列名。塩基長も書いてある。次の>までがde novoアセンブリにより作られた塩基配列である。この様なファイル形式をfasta形式とよぶ。余談だが、fasta形式のファイルには1本の塩基配列中に改行がある場合とない場合があり、今回は改行が含まれている。”fasta formatter”というツールを使うと、配列途中の改行を取り除くことができる。こうすると、wc -lコマンドで配列数を調べることが可能だ。では、このファイルにどれぐらいの塩基長の配列が何個ぐらい含まれているのか統計をとってみる。cd-hit-estというサイトが使いやすい。次のリンク先にアクセスし、cd-hit-estというタブをクリックする。http://weizhong-lab.ucsd.edu/cdhit_suite/cgi-bin/index.cgi
”ファイルを選択”ボタンをクリックしてTrinity.fastaを選択し、下の”Submit”ボタンをクリックすればよい。しばらくすると、いくつかのファイルを選べるようになる。そのうち、xxxx.fas.0.statというファイルをクリックすると、以下のように表示される。
Sequence type DNA (以下省略) |
この結果を見ると、配列の数は6866本と少なく、そのうち77%は399塩基より短い。平均長は363塩基しかない。今回は、計算時間を短くするために、headコマンドによりリード数を減らした。これがmRNAらしい長い配列が多くできなかい理由の一つだ。ちなみに、headコマンドによる減数処理を行っていないfastqファイルを使ってここまで同様の操作を実行してみると、得られる配列数が82492と大幅に増えた。平均長は536塩基と大きくなったとはいえ、植物のmRNAとしてはまだまだ短い。もともとのサンプルの質自体も、もしかしたら良くなかったのかもしれない(例えば抽出したRNAサンプルが分解していた)。
(2-3-2) Velvetによるde novoアセンブリ
(今回の実習ではたぶん時間がたりなくなるのでこの項目は省略。最後に余裕があればやります)
Velvetも試してみよう。mRNAのアセンブリの場合は、velveth, velvetg、oasesという3つのツールを順に使用する。まずvelveth。k-merという変数を調整しながら解析をするとよいが、以下の条件でまずは実行してみる。簡単にオプションの説明をしておくと、”velveth”に続く、”~/work/sakito/”は結果をこのディレクトリに出力すること、65はk-merを65にすることを指定している。k-merの意味は、似ているリード同士を探すときに、k-merで指定した塩基長でリード配列を区切って比較する、というものだそうだ。k-merを大きくするとアセンブルを厳しくし、小さくすると甘くする。今回はインストールの際にk-merの上限値を99としたので、99以下の奇数を指定できる。-separate、-shortPaired、-fastqはそれぞれ、入力ファイルが、paired endの2つの配列ファイルに分かれていること、paiered endの短い配列であることを、fastq形式であることを指定している。入力ファイルの置き場所を指定するときは、”~”を使うパスは使えないようだ。下の入力例には”/manager”という文字が書いてあるが、今回使用する本学のシステムの固有の事情によりこうなる。自分のPCにBio-Linux 8の場合は、この文字は不要のはずだ。sakito/の部分は、ユーザーアカウントごとに異なるので各自修正しなくてはならない。
kitajima@kitajima[kitajima] ~/_Tools/velvet/velveth ~/work/sakito/ 65 -separate -shortPaired -fastq /home/manager/work/sakito/ERR260307_1.fastq /home/manager/work/sakito/ERR260307_2.fastq |
実行すると以下のようになるはずだ。Trinityに比べると一瞬で終わる。
kitajima@kitajima[kitajima] ~/_Tools/velvet/velveth ~/work/sakito/ 65 -separate -shortPaired -fastq /home/manager/work/sakito/ERR260307_1.fastq /home/manager/work/sakito/ERR260307_2.fastq [0.000002] Reading FastQ file /home/manager/work/sakito/ERR260307_1.fastq; [0.002793] Reading FastQ file /home/manager/work/sakito/ERR260307_2.fastq; (中略) [5.752991] Destroying splay table [5.771746] Splay table destroyed kitajima@kitajima[kitajima] |
次にvelvetgを実行する。~/work/sakito/は結果の出力先をこのディレクトリに指定している。-cov_cutoffは、カバレージ(リードがどのくらい張り付いたかを示す)の低い配列を除外するオプションで、とりあえずautoとしておく。-ins_lengthはシーケンスに供したcDNA断片の長さで、今回のサンプルでは300である。-ins_length_sdはその標準偏差で、今回は不明なので適当に100としておく。-read_trkgは、リードトラッキングなるものを実行することを指定する(あまり大事ではないようなので指定しなくても良いのかもしれない)。
kitajima@kitajima[kitajima] ~/_Tools/velvet/velvetg ~/work/sakito/ -read_trkg yes -cov_cutoff auto -ins_length 300 -ins_length_sd 100 -exp_cov auto |
実行すると、以下のように表示されるはずだ。画面の表示の読み方はよく分からないが、推定カバレージが11.851964、推定カバレージカットオフは5.925982とのことなので、”-cov_cutoff”を設定する場合はこれを目安にすることになると思う。
kitajima@kitajima[kitajima] ~/_Tools/velvet/velvetg ~/work/sakito/ -read_trkg yes -cov_cutoff auto -ins_length 300 -ins_length_sd 100 -exp_cov auto [0.000000] Reading roadmap file /home/kitajima//Roadmaps (中略) [8.069185] Estimated Coverage = 11.851964 [8.069205] Estimated Coverage cutoff = 5.925982 Final graph has 679 nodes and n50 of 296, max 1475, total 190083, using 176587/600000 reads kitajima@kitajima[kitajima] |
最後にoasesを実行する。オプションの~/は、結果の出力先をホームディレクトリに指定している。-min_trans_lgthは、出力する配列の長さの下限を指定する。今回は200に設定しておくが、通常はもっと大きな値(例えば400)でもよいとおもう。-ins_lengthは、velvetgの場合と同じで、今回のサンプルでは300である。-cov_cutoffは、6としておく。カバレージがそれより低い配列は除去される。
kitajima@kitajima[kitajima] ~/_Tools/oases/oases ~/ -ins_length 300 -min_trans_lgth 200 -cov_cutoff 6 |
実行すると、以下のように表示されるはずだ。
kitajima@kitajima[kitajima] ~/_Tools/oases//oases ~/ -ins_length 300 -min_trans_lgth 200 -cov_cutoff 6 [0.000000] Reading graph file /home/kitajima//Graph2 (中略) [1.037848] Exporting transcripts to /home/kitajima//transcripts.fa [1.047910] Exporting transcript contigs to /home/kitajima//contig-ordering.txt [1.054493] Finished extracting transcripts, used 175596/600000 reads on 646 loci kitajima@kitajima[kitajima] |
アセンブリの結果は、ホームディレクトリの”transcripts.fa”というファイルに出力された。Trinityのときと同様にcd-hit-estで調べると、得られた配列は628本、平均長366で、200から399塩基長の配列が全体の77%を占めている。
ちなみに、headコマンドによる減数処理を行っていないfastqファイルを使ってここまで同様の操作(ただしoasesの-cov_cutoffを12に変更した)を実行してみると、得られる配列数が15673と大幅に増えた。平均長は512塩基と大きくなったとはいえ、植物のmRNAとしてはまだまだ短い。
今回は、k-merの値を1つに固定して解析した。oasesのフォルダの中に、oases_pipeline.pyというファイルがある。これは、pythonというプログラミング言語のスクリプトで、これを実行すると、指定する範囲のさまざまなk-merで順に解析したあと、結果を1つにまとめて重複する配列を除去してくれる。oasesの説明書に使い方が書いてあるので、興味のあるひとは試してみるとよい。ただし、このpythonスクリプトをテキストエディタで開いて、velveth, velvetg, oasesの置き場所を記述した部分をすべて自分の環境に合わせて書き換える必要がある。例えば、今回のシステムの場合(固有の事情によりやや変わったパスになっている)、
subprocess.Popen(['velveth → subprocess.Popen([/home/manager/_Tools/velvet/velveth
置き場所は、先述のとおり、ファイルマネージャで当該のファイルを右クリックしてプロパティを選べば表示される。
あらかじめパスを通しておけば、たぶんその必要はない。
(2-4) Bowtie2によるマッピング
推定mRNA配列データベースができあがったので、これをリファレンス配列としてリード(ホームディレクトリのERR260307_short_1.fastqおよびERR260307_short_2.fastq)を張り付けてみる。Trinityとvelvetで作成した2セットのアセンブリがあるが、以降はTrinityで作成した方を使用することにする。マッピングにはbowtieのver.2をつかう。このツールは、fastqファイルの各リードが、どのリファレンス配列のどの位置に一致するかを調べる。リファレンス配列としたいfastaファイルは、まずbowtie2-buildというツールを使って専用のインデックスファイルに変換する。書式は、”bowtie2-build 元のfastaファイル インデックス名”だ。インデックスには適当な名前をつければよいが、ここではTrinityFastaLilyとしておく。
kitajima@kitajima[kitajima] bowtie2-build ~/work/sakito/Trinity_sample/Trinity.fasta ~/work/sakito/TrinityFastaLily Settings: Output files: "TrinityFastaLily.*.bt2" Line rate: 6 (line is 64 bytes) Lines per side: 1 (side is 64 bytes) (中略) ebwtTotSz: 832256 color: 0 reverse: 1 Total time for backward call to driver() for mirror index: 00:00:01 kitajima@kitajima[kitajima] |
~/work/sakitoディレクトリにはTrinityFastaLilyほにゃらら.bt2というファイルがいくつかできたのが確認できるだろう。つづいてマッピングする。次のような書式でタイプする。”bowtie2 オプション -x インデックス FASTQファイル 出力ファイル”。オプションとしては、とりあえず以下のものをよく使用すると思う。
--un-conc filename ; マップされなかったpaired endのリードを指定するfastqファイルに出力する。
(ちなみに、入力ファイルがpaired endでない、つまり一方向のリードの場合は、--un filename ファイル名と指定する)
-p 使用するCPUのスレッド数
-1 query1.fastq -2 query2.fastq ; paired endの2つの入力ファイルを指定する。
-X 数字 pair-endの最大インサート長さ。シーケンスに供したcDNA断片の最大サイズのことで、デフォルト値は500とのことなので、今回は指定しない。
-L 数字 対象配列と最初に比較するときのリードのタネ長さ(適切な表現が分からない。英語では”length of seed substring”と呼ぶ)で、アラインのパラメータで感度と速さに関わる。今回のリードは長さが60塩基以上なので設定可能な最大値の32としておく。
-S ファイル名 マップの結果の出力先。sam形式ファイルとするよう指定している。
では、やってみる。
kitajima@kitajima[kitajima] bowtie2 -p 2 -L 32 -x ~/work/sakito/TrinityFastaLily -1 ~/work/sakito/ERR260307_short_1.fastq -2 ~/work/sakito/ERR260307_short_2.fastq -S ~/work/sakito/bowtie2result.sam --un-conc ~/work/sakito/out_unmapped.fastq |
しばらく待つと以下のように表示される。
kitajima@kitajima[kitajima] bowtie2 -p 2 -L 32 -x ~/work/sakito/TrinityFastaLily -1 ~/work/sakito/ERR260307_short_1.fastq -2 ~/work/sakito/ERR260307_short_2.fastq -S ~/work/sakito/bowtie2result.sam --un-conc ~/work/sakito/out_unmapped.fastq 300000 reads; of these: 300000 (100.00%) were paired; of these: 97242 (32.41%) aligned concordantly 0 times 51439 (17.15%) aligned concordantly exactly 1 time 151319 (50.44%) aligned concordantly >1 times ---- 97242 pairs aligned concordantly 0 times; of these: 1196 (1.23%) aligned discordantly 1 time ---- 96046 pairs aligned 0 times concordantly or discordantly; of these: 192092 mates make up the pairs; of these: 169316 (88.14%) aligned 0 times 12766 (6.65%) aligned exactly 1 time 10010 (5.21%) aligned >1 times 71.78% overall alignment rate kitajima@kitajima[kitajima] |
全リードの71.78%がうまくマッピングできた。良好な成績と言える。マッピングの結果はbowtie2result.samという名前のファイルに書き込まれている。マッピングされなかったリードは、ホームディレクトリのout_unmapped.1.fastqおよびout_unmapped.2.fastqの2つのファイルに出力された。
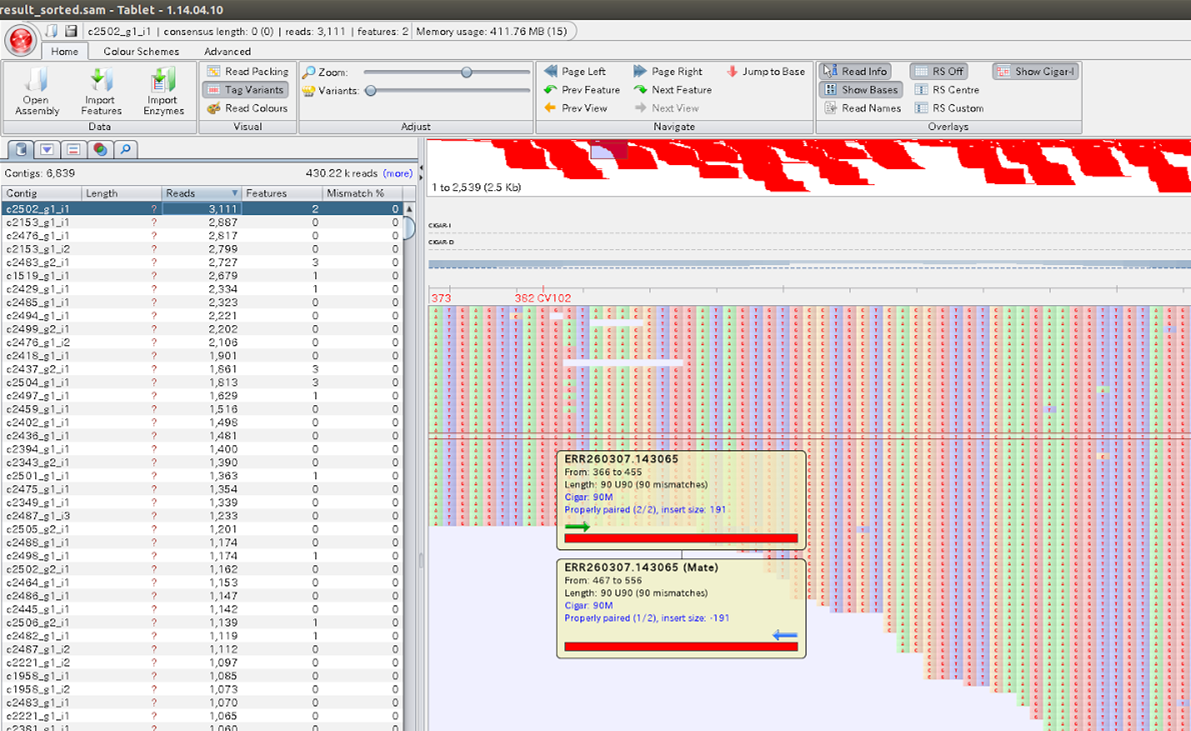
bowtie2の結果を、samtoolsというデータ変換ツールですこしばかりの前処理をすると、Tabletというアプリを使って可視化することができる。細かい話は抜きにして、次のように順番にタイプすると良い。
kitajima@kitajima[kitajima] samtools view -bS ~/work/sakito/bowtie2result.sam > ~/work/sakito/bowtie2result.bam [samopen] SAM header is present: 6860 sequences. kitajima@kitajima[kitajima] samtools sort ~/work/sakito/bowtie2result.bam ~/work/sakito/bowtie2result_sorted kitajima@kitajima[kitajima] samtools view -h ~/work/sakito/bowtie2result_sorted.bam > ~/work/sakito/bowtie2result_sorted.sam kitajima@kitajima[kitajima] tablet ~/work/sakito/bowtie2result_sorted.sam |
Tabletの使い方は、実際に使ってみればすぐ分かるとおもう。samtoolsについては、以下のサイトが参考になる。
https://cell-innovation.nig.ac.jp/wiki/tiki-index.php?page=samtools
Tablet画面の左側の表には、リファレンス配列ごとに張り付いたリードの数が表示されている。おおざっぱに言えば、これが発現ランキングということになるが、リファレンス配列の塩基長がばらばらなので公平とはいえない。例えば、リファレンス配列すなわちmRNAの分子数が同じだったとしても、張り付いたリードの数は、2kbのmRNAでは1kbのmRNAの場合の2倍の大きさになってしまう。遺伝子間での発現の強弱を比較したければ、塩基長あたりのリード数で比較すべきた。
(2-5) RPKMの計算
RPKMは、Reads Per Kilobase of exon per Million mapped readsの略で、リードの出現回数を塩基長とリード総数で割って、1kb当たりと100万リード当たりに標準化したものだ。このように標準化すると、長さの異なる遺伝子の間で発現強度を比較可能になり、さらに別の実験サンプルの結果との比較も可能になる。以下のようにして、RPKMに基づく発現ランキング表を作ることにする。そのためには、各リファレンス配列の塩基長と、各リファレンスに張り付いたリードの数をしらべなくてはならない。いろいろなLinuxのコマンドを使うが、細かい解説はしない。コマンドの意味や使い方に興味のあるひとは、例えば、”linux grep 使い方”というようなキーワードでググってみるとよい。最初に、bowtie2が出力したsamファイル(bowtie2result.sam)に何が書いてあるのか調べてみよう。冒頭20行をheadコマンドで見てみる。
kitajima@kitajima[kitajima] head -20 ~/work/sakito/bowtie2result.sam @HD VN:1.0 SO:unsorted @SQ SN:c0_g1_i1 LN:263 @SQ SN:c1_g1_i1 LN:807 (中略) @SQ SN:c17_g1_i2 LN:461 @SQ SN:c18_g1_i1 LN:1327 @SQ SN:c20_g1_i1 LN:852 kitajima@kitajima[kitajima] |
左から順番に、“@SQ”、タブ区切り、”SN”、”:”、続いてリファレンス配列の名称、タブ区切り、”LN”、”:”、塩基長と続く。この様にsamファイルの冒頭には、リファレンス配列の情報が書いてある。マッピングの結果は、もっと下に書いてある。ファイルの末尾20行を見てみよう。headコマンドの代わりにtailというコマンドを使う。
kitajima@kitajima[kitajima] tail -20 ~/work/sakito/bowtie2result.sam ERR260307.299992 77 * 0 0 * * 0 0 AGCAGTACCTCGACGAGAAGACCATCTTCCACCTCAACCCATCGGGTCGCTTTGTCATTGGTGGTCCTCATGGTGATGCTGGGCTTACTG ?@@DDAABFHHHHGGIIFE;F@9BDFGIIIIIIGIID@GGGHICEFGB8BECB>B;;AB@C6=?33>@C>>@C3>>>@>:@@8198:A>: YT:Z:UP ERR260307.299992 141 * 0 0 * * 0 0 GGTCGACCTTGGTGGGGTCCTTACCGGAGAAGGCACCACCACCGTGAGCTCCCCATCCTCCATAGGTGTCGATGATAATTTTACGACCAG @?=DD@:AF<DAFHHIH@EECH4?CCFGGGF;B(8BBF2;FHI4CEFE=;@??A@>6;@@CDC;;>5>(,8A?CCCCC::AC>48?59@? YT:Z:UP ERR260307.299993 73 c1642_g1_i1 250 26 90M = 250 0 ATCCCCACATCCTCATCTATGTGACTTCAATTCTAATTTCCATGTATTCAACACCCAATTGGGCAAAGAAGAATCGCGATGGCGGGAGAC @@<DDFFFBCFHFIG@E@HAEAEHGIIGIHECHIIIIIFIIGBHGGIGIEEHGHFHFHDFGIGIIGCHIIICGHHEEB############ AS:i:-5 XS:i:-44 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:17C72 YT:Z:UP (以下略) |
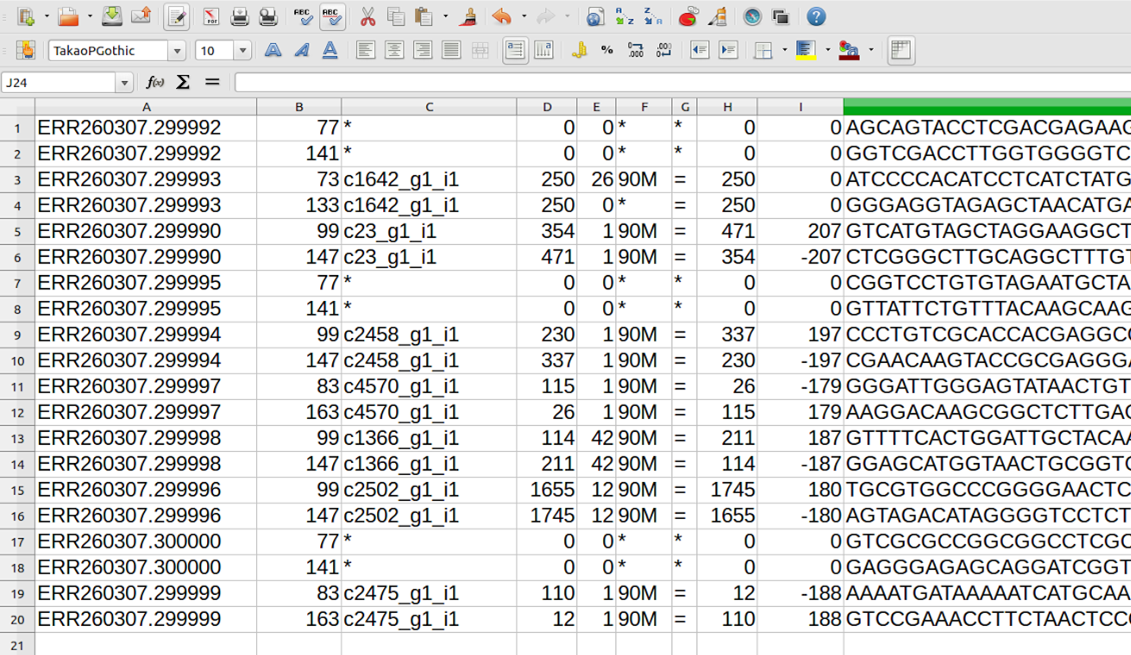
分かりにくいので、tailの実行結果を、画面の代わりにファイルに出力させてLibrOffice calcで見てみる。ファイルに出力させるには、tailコマンドの末尾に”> 出力先ファイル名”と書き加える。出力先ファイルの名前はなんでもよい。LibreOffice calcでファイルを読み込むときに区切り方法をきかれるので、”タブ区切り”にチェックをいれ、他はオフにする。
各行の構成がわかりやすくなった。タブ区切りで、各行の1番めのフィールド(例;ERR260307.299993)がリードの名称、3番めのフィールドが貼り付き先のリファレンス配列名(例;c1642_g1_i1)となっている。また、リードがどのリファレンス配列にもマップされなかった場合には、第3フィールドに*と書いてある。
ではまず、冒頭部分から各リファレンス配列の長さを抽出し、RefLen.csvという名前のファイルに書き出す。その前に、ファイルの1行めに各フィールドの名前を書いておこう。以下のようにタイプする。”reference”タブ区切り”Length”と出力してホームディレクトリのReflen.csvというファイルに書き込め、という意味だ。
kitajima@kitajima[kitajima] echo -e "reference""\t""length" > ~/work/sakito/RefLen.csv |
次に、以下のようにタイプする。コマンドの概略を述べると、当該の行では第1フィールドが必ず@SQなので、これを目印にしてgrepコマンドでbowtie2result.samから当該の行を抜きだす。次にsedコマンドで”:”をタブ区切りに置換し、cutコマンドで第3(リファレンス配列名)および第5フィールド(塩基長)を切り出し、結果をホームディレクトリのRefLen.csvというファイルの末尾に追記する。”|”はパイプと呼ばれるもので、”|”の前のコマンドの実行結果を”|”の後のコマンドに渡す、という意味である。>>は、この記号の左側の実行結果を右側のファイルの末尾に追記すると言う意味だ。それぞれのコマンドの実行結果を見てみたければ、パイプで繋がず、代わりにモニタかファイルに出力してみるとよい。最後に、wc -lコマンドで行数を、headコマンドで出来上がったファイルの内容を確認しておく。
kitajima@kitajima[kitajima] grep "@SQ" ~/work/sakito/bowtie2result.sam | sed -e 's/:/\t/g' | cut -f3,5 >> ~/work/sakito/RefLen.csv kitajima@kitajima[kitajima] wc -l ~/work/sakito/RefLen.csv 6861 /home/manager/work/sakito/RefLen.csv kitajima@kitajima[kitajima] head -10 ~/work/sakito/RefLen.csv reference length c0_g1_i1 263 c1_g1_i1 807 c3_g1_i1 426 c4_g1_i1 406 c5_g1_i1 238 c6_g1_i1 413 c6_g1_i2 463 c8_g1_i1 383 c9_g1_i1 554 kitajima@kitajima[kitajima] |
マッピングの集計結果は、ReadNum.csvというファイルに書き込むことにする。上と同様にechoコマンドで1行めにフィールド名をあらかじめ記入しておく。
kitajima@kitajima[kitajima] echo -e "reference""\t""Number" > ~/work/sakito/ReadNum.csv |
次に、以下のようにタイプして、マッピング結果のデータを取り出す。コマンドの概略を述べると、bowtie2result.samの内容をcatコマンドで読み出してgrep -vコマンドに引き渡し、"@SQ"という文字を含む冒頭の行を削除し、awkのprintコマンドによりその結果の第3フィールド(つまりマップされたリファレンス配列名)を切り出し、さらにgrep -vコマンドで”*”を含む行を削除して、その結果をホームディレクトリのReadTemporary.csvというファイルに書き出す。
kitajima@kitajima[kitajima] cat ~/work/sakito/bowtie2result.sam | grep -v "@SQ" | awk '{print $3}' | grep -v "*" > ~/work/sakito/ReadTemporary.csv |
ReadTemporary.csvの冒頭部分を一応見ておこう。
kitajima@kitajima[kitajima] head -10 ~/work/sakito/ReadTemporary.csv SO:unsorted PN:bowtie2 c2374_g1_i1 c2374_g1_i1 c2437_g2_i1 c2437_g2_i1 c2399_g1_i1 c2399_g1_i1 c2485_g1_i1 c2485_g1_i1 kitajima@kitajima[kitajima] |
冒頭の2行はリファレンス配列名ではない。邪魔なので、sed -eコマンドを使って、削除しておこう。ひとつ前のステップからやり直す。
kitajima@kitajima[kitajima] cat ~/work/sakito/bowtie2result.sam |grep -v "@SQ" | awk '{print $3}' | grep -v "*" | sed -e '1,2d' > ~/work/sakito/ReadTemporary.csv kitajima@kitajima[kitajima] head -10 ~/work/sakito/ReadTemporary.csv c2374_g1_i1 c2374_g1_i1 c2437_g2_i1 c2437_g2_i1 c2399_g1_i1 c2399_g1_i1 c2485_g1_i1 c2485_g1_i1 c3439_g1_i1 c3439_g1_i1 kitajima@kitajima[kitajima] |
うまくいった。tail -10コマンドでファイルの末尾も確認するとよい。最後に、このファイルに各リファレンス配列名が何回出現するかを数えれば良い。以下のようにタイプする。意味は、sortコマンドによりReadTemporary.csvの行をアルファベット順に並べ替え、uniq -cコマンドにより同じ行(つまり同じリファレンス配列名)が連続して出現した回数と、そのリファレンス配列名を出力する。awkのprintコマンドで、出現回数とリファレンス配列名の順序を入れ替えて、タブ区切りとし、その結果をホームディレクトリの先ほど作成したReadNum.csvというファイルの末尾に追記する。headコマンドで中身を確認しておく。
kitajima@kitajima[kitajima] cat ~/work/sakito/ReadTemporary.csv | sort | uniq -c | awk '{print $2"\t"$1}' >> ~/work/sakito/ReadNum.csv kitajima@kitajima[kitajima] head -10 ~/work/sakito/ReadNum.csv reference Number c0_g1_i1 20 c1000_g1_i1 26 c1001_g1_i1 90 c1003_g1_i1 28 c1004_g1_i1 46 c1005_g1_i1 58 c1008_g1_i1 22 c1009_g1_i1 16 c1010_g1_i1 16 kitajima@kitajima[kitajima] wc -l ~/work/sakito/ReadNum.csv 6832 /home/manager/work/sakito/ReadNum.csv kitajima@kitajima[kitajima] |
wc -lコマンドで行数を調べると68432、タイトル行を除くと6831行で、もとのリファレンス配列の数より少ない。これは、リードが1本も張り付かなかったリファレンス配列がここには含まれていないためだ。
では次に、このようにして作成したRefLen.csvとReadNum.csvを連結しよう。2つの表の連結は、Rというプログラムでやってみる。端末に、Rとタイプすると起動する。”>”と表示されれば入力待ち状態だ。
kitajima@kitajima[kitajima] R R version 3.1.1 (2014-07-10) -- "Sock it to Me" (中略) 'help()' とすればオンラインヘルプが出ます。 'help.start()' で HTML ブラウザによるヘルプがみられます。 'q()' と入力すれば R を終了します。 > |
以下のように順にタイプしていく。概略を述べると、1行めと2行めでは作成した2つのファイルを読み込み、それぞれ変数LとNに入力する。”header=T”はファイルの1行目がタイトル行であることを、 “sep="\t"”はタブ区切りであることをRに教えている。3行めでは2つのファイルを、リファレンス配列が一致するように連結して、変数Mに入力する。”all=T”は、2つのファイルの一方にしか存在しないリファレンス配列名があった場合には、欠損している方にNAの文字を加えることを指示している。今回は、ReadNum.csvに欠損がある(1本もマッピングされなかったリファレンス配列があるため)ので、”Number”のフィールドにNAの文字を入れてくれる。
> L <- read.csv("~/work/sakito/RefLen.csv", header=T, sep="\t") > N <- read.csv("~/work/sakito/ReadNum.csv", header=T, sep="\t") > M <- merge(L, N, all=T) |
どんな表ができたか確認のため、変数Mの冒頭を見ておこう。head(M)とタイプする。
> head(M) reference length Number 1 c0_g1_i1 263 20 2 c1000_g1_i1 218 26 3 c1001_g1_i1 454 90 4 c1003_g1_i1 261 28 5 c1004_g1_i1 461 46 6 c1005_g1_i1 563 58 > |
問題がなければ、結果をホームディレクトリのReadNum_Len.csvというファイルに出力して、Rを終了する。”row.names=FALSE”は、行番号を出力しない、という意味である。
> write.table(M, file="~/work/sakito/ReadNum_Len.csv", row.names=FALSE, sep="\t") > q() Save workspace image? [y/n/c]: n kitajima@kitajima[kitajima] |
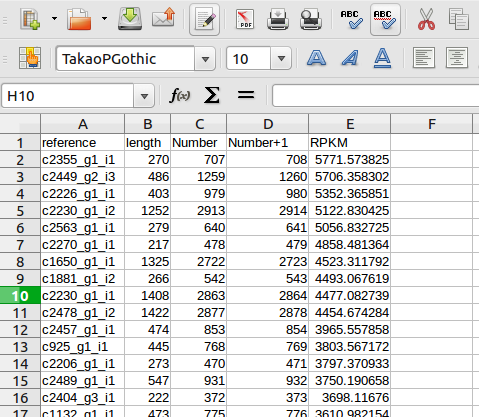
では最後に、ReadNum_Len.csvをLibreOffice calcで読み込んでみよう。これ以降の操作は、Excelとほぼ同じだ。計算をする前に、ひとつやらなくてはならないことがある。表を下にスクロールすると、張り付いたリード数すなわち”Number”の列が数字ではなく、”NA”となっているところがある。これは、データ欠損、つまりこのリファレンス配列にはリードが1本も張り付かなかった、ということだ。NAの文字列を検索してすべて0に置換しておこう。今回は行わないが、他のmRNA-seqの結果と比較する際にはしばしばlog表記する。データに0があるとlogをとれないので、すべてのデータに1を加えておく。前処理はここまでで、いよいよRPKMを計算する。RPKMの計算式は、自分で考えられるだろう。リード総数の算出法は、異論があるかもしれないが、ここでは表の[リード数+1]を上から下まですべて足した値としておく。RPKMの順にソートすると、右のような結果になった。
ちなみに、LibreOffice calcで行った最後の処理も、Rの中ですることが可能だ。もういちどRを起動して、以下のようにタイプする。Rで先ほど行った処理に加えて、(1)では、変数M(つまりmerge関数で作った表)の中の”NA”を0に置き換える。(2)では、変数MのNumberとなづけた列(つまりリード数)に1を加えて、結果を変数MのNumPlus1という列に書き込む。(3)では、RPKMを計算して、変数MのRPKMという名前の列に書き込む。sum()は合計を計算する関数だ。最後に、変数MをReadNum_Len_RPKM.csvというファイルに出力する。下には書いてないが、(1)~(3)の各ステップを実行するごとに、確認のためhead(M)とタイプしてみるとよい。
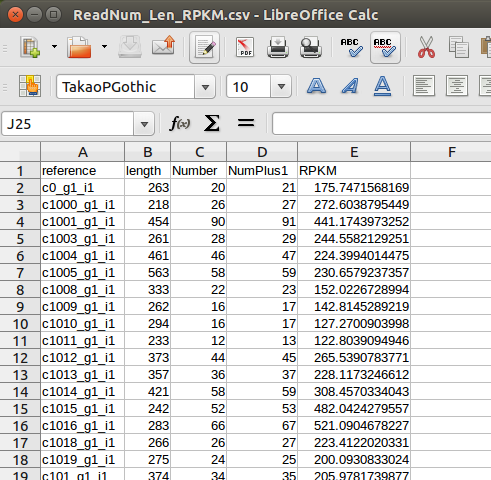
> L <- read.csv("~/work/sakito/RefLen.csv", header=T, sep="\t") > N <- read.csv("~/work/sakito/ReadNum.csv", header=T, sep="\t") > M <- merge(L, N, all=T) > M[is.na(M)] <- 0 > M$NumPlus1 <- M$Number + 1 > M$RPKM <- M$NumPlus1/sum(M$NumPlus1)/M$length*1000*1000000 > write.table(M, file="~/work/sakito/ReadNum_Len_RPKM.csv", row.names=FALSE, sep="\t") > q() Save workspace image? [y/n/c]: n kitajima@kitajima[kitajima] | (1) (2) (3) |
出力したファイルReadNum_Len_RPKM.csvをLibreOffice calcで開くと、右のようになっている。
だいぶ煩雑な作業だった。しかし、bowtie2以降に関して言えば完全な定型処理だ。シェルスクリプトを書けば、ここまでの一連の操作をコマンド一発で実行できるようになる。下に演習課題として挙げておくので、明日シェルスクリプトを学んだら、やってみるとよい。
(2-6) データベースにmRNA配列がいくつか登録されている場合のアセンブリについて。
今回は、公開されているmRNA配列がまったく無いという想定の下に、完全なde novoアセンブリを行った。しかし大抵の場合は、その生物のmRNA配列が少しぐらいはデータベースに公開されているだろう。これらをどう活用すればもっとも有効なのだろうか。筆者は、bowtie2を使って、既知のmRNAに対応するリードをあらかじめ除去してからde novoアセンブリを行えばよいと考えている。bowtie2には、マップされなかったリードをfastqファイルに出力するオプションがあることをすでに紹介した。そうして得られた推定mRNA配列のファイルを、既知mRNA配列のファイルと連結して1つのfastaファイルにまとめればよい。リボソームRNAの混入が問題になる場合も同じ方法が可能なはずだ。
演習課題1 bowtie2が出力したマッピング結果のファイルを使ってRPKMを計算する作業を、今回は1行ずつ手作業で行った。この作業をシェルスクリプトで記述して動作させなさい。 |
3. モデル生物のmRNA-seq解析 |
(3-1) データの準備
(3-1-1) ゲノム配列のダウンロード
例えば、酵母、アラビドプシス、ヒト、ショウジョウバエなどの場合は、ゲノム配列が解読されていて、染色体のどの領域が遺伝子か、どこがエクソンか、というアノテーション情報が公開されている。そのような生物種のmRNA-seq解析では、アノテーション付きゲノムデータをリファレンス配列としてマッピングすればよい。マッピングツールとしてはTopHat2を使う。TopHat2は、内部でbowtie2を使っているが、リードが2つのエクソンをまたいでいてもうまくマッピングしてくれる。まずは、リファレンス配列として使うゲノム情報を入手する。以下のリンク先にillumina社が提供している各モデル生物のデータがある。
http://cufflinks.cbcb.umd.edu/igenomes.html
今回はアラビドプシスを使ってみる。アラビドプシスのEnsamblのTAIR10をクリックすると、ダウンロードディレクトリに、Arabidopsis_thaliana_Ensembl_TAIR10.tar.gzという圧縮ファイルをダウンロードするはずだ。これを展開するとArabidopsis_thaliana_Ensembl_TAIR10という名前のディレクトリができるので、ホームディレクトリに移す。中をみてみると、いろいろなファイルが入っている。これから使うのは、1つめが、EnsemblフォルダのTAIR10ファルダのAnnotationフォルダのGenesフォルダにあるgenes.gtfファイルだ。これがアノテーション情報である。TopHat2用のインデックスファイルも用意されていて、TAIR10フォルダのSequenceフォルダのBowtie2Indexの中に入っている。もし、TopHat2の内部でbowtie2のかわりにbowtie1を使いたい場合は、bowtieIndexフォルダの中に入っているインデックスファイルを使用する。本来なら、bowtie2を使うのだが、執筆時点でどういうわけかbowtie2インデックスをTophat2がうまく認識しないので、今回はbowtie1とそのインデックスを使う。後ほど改めて説明する。
(3-1-2) アラビドプシスのリードデータの用意
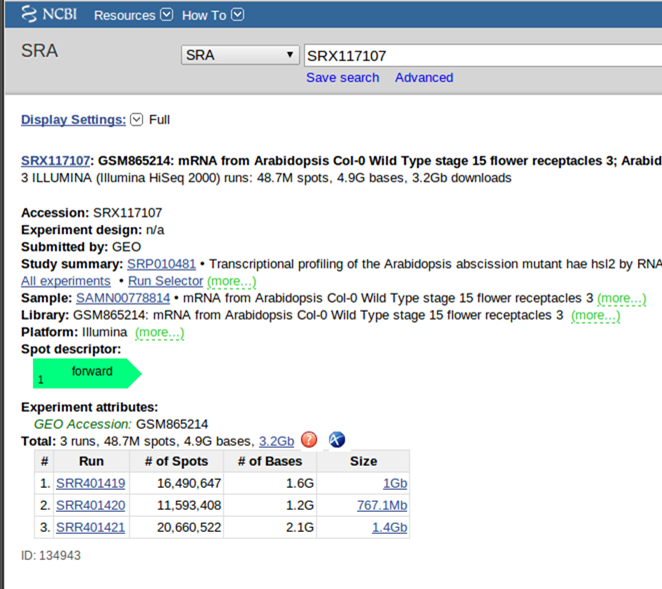
ユリのときと同じように、NCBIのSRAデータベースを検索すると多数みつかる。テッポウユリのデータと同様にpaired-endのデータもあるが、ざっとみるとsingle endのものが多いようだ。single endというのは、高速シーケンサーでcDNA断片を読むとき、両末端のどちらか一方だけを読む手法である。今回はsingle endのデータを使ってみよう。アクセッションNo. SRX117107 (http://www.ncbi.nlm.nih.gov/sra/?term=SRX117107)を見てみる。
説明文によると、材料はアラビドプシス(Arabidopsis thaliana)、 エコタイプcol-0の花托(花の基部のふくらんだところ)より抽出したmRNAで、シーケンシングにはillumina社HiSeq 2000を使用したとのことだ。3回の繰り返し実験を行ったので3つのsraデータ(SRR401419.sra SRR401420.sra SRR401421.sra)が登録されている。これらをすべてダウンロードして、ホームディレクトリに移す。以降は、とりあえずSRR401419.sraだけを先に進める。テッポウユリのときと同様に、まず、sratoolskitのfastq-dumpでfastqファイルに変換する。single endでの実験なので、テッポウユリのときに付け加えたオプションの”--split-files”はなくてもよいが、付け加えておくと、*_1.fastqという名前のファイルを作る。
kitajima@kitajima[kitajima] ~/_Tools/sratoolkit/bin/fastq-dump ~/work/sakito/SRR401419.sra --split-files -O ~/work/sakito |
次のように表示されれば成功である。
kitajima@kitajima[kitajima] ~/_Tools/sratoolkit/bin/fastq-dump ~/work/sakito/SRR401419.sra --split-files -O ~/work/sakito Read 16490647 spots for /home/manager/work/sakito/SRR401419.sra Written 16490647 spots for /home/manager/work/sakito/SRR401419.sra kitajima@kitajima[kitajima] |
ファイルマネージャで~/work/sakitoディレクトリを見ると、SRR401419_1.fastqという名前の5.4GBのファイルができている。前回とおなじく、計算時間を短くするために、headコマンドで最初の30万リード、すなわち最初の120万行だけを取り出してサブセットのリードデータをつくろう。 wc -lコマンドで120万行であることが確認できればOKだ。
kitajima@kitajima[kitajima] head -1200000 ~/work/sakito/SRR401419_1.fastq > ~/work/sakito/SRR401419_short_1.fastq kitajima@kitajima[kitajima] wc -l ~/work/sakito/SRR401419_short_1.fastq 1200000 /home/manager/work/sakito/SRR401419_short_1.fastq kitajima@kitajima[kitajima] |
(3-2) TopHat2によるマッピング
作成されたファイル(SRR401419_short_1.fastq)を、TopHat2を用いて、アラビドプシスのゲノム配列にマッピングする。tophat2 -helpとタイプすると書式等の情報が見られる。とりあえずは、次のようにタイプすれば良い。
tophat2 -p CPUスレッド数 -G gtfファイル名 -o 出力先ディレクトリ bowtie2_index fastqファイル
結果の出力先は、~/work/sakito/tophatresult_SRR401419 とする。すでに述べたとおり、Tophat2は内部でbowtie2を使用する。しかし、オプションを指定すればbowtie1を使わせることもできる。本来ならbowtie2を使いたいので、以下のようにタイプするべきだ。
tophat2 -p 3 -G ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf -o ~/work/sakito/tophatresult_SRR401419 ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/Bowtie2Index/genome ~/work/sakito/SRR401419_short_1.fastq |
しかし、テキスト執筆時点(2014.08.06)でTophat2がうまくbowtie2のインデックスを見つけてくれない。理由はわからない。今回は、暫定的措置として、bowtie1を使用させる。bowtie2を使用する場合に比べて、オプションに、”--bowtie1”を追加し、インデックスファイルの場所を、”~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/BowtieIndex/genome”と変更する。以下のようにタイプすれば良い。
kitajima@kitajima[kitajima] tophat2 --bowtie1 -p 3 -G ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf -o ~/work/sakito/tophatresult_SRR401419 ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/BowtieIndex/genome ~/work/sakito/SRR401419_short_1.fastq [2014-09-05 16:27:23] Beginning TopHat run (v2.0.9) ----------------------------------------------- [2014-09-05 16:27:23] Checking for Bowtie Bowtie version: 1.0.1.0 [2014-09-05 16:27:23] Checking for Samtools Samtools version: 0.1.19.0 (中略) [2014-09-05 16:56:40] A summary of the alignment counts can be found in /home/manager/work/sakito/tophatresult_SRR401419/align_summary.txt [2014-09-05 16:56:40] Run complete: 00:29:17 elapsed kitajima@kitajima[kitajima] |
結果は、指定したtophatresult_SRR401419ディレクトリのaccepted_hits.bamというファイルに書き込まれている。
(3-3) CufflinksによるFPKMの計算
このファイルをcufflinksというツールに読み込ませて張り付いたリード数を集計するのだが、そのまえに”samtools sort”というツールを使って、データの前処理をする。以下のようにタイプする。
kitajima@kitajima[kitajima] samtools sort ~/work/sakito/tophatresult_SRR401419/accepted_hits.bam ~/work/sakito/tophatresult_SRR401419/sort_accepted_hits kitajima@kitajima[kitajima] |
tophatresult_SRR401419ディレクトリにsort_accepted_hits.bamという名前のファイルができたはずだ。これをcufflinksに読ませる。次のような書式で入力すれば良い。
cufflinks -o 出力先ディレクトリ -p CPUのスレッド数 -G gtfファイル名 入力ファイル(bam)
今回は次のようにタイプする。出力先は、tophatresult_SRR401419ディレクトリとしておく。
kitajima@kitajima[kitajima] cufflinks -o ~/work/sakito/tophatresult_SRR401419/ -p 3 -G ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf ~/work/sakito/tophatresult_SRR401419/sort_accepted_hits.bam Warning: Your version of Cufflinks is not up-to-date. It is recommended that you upgrade to Cufflinks v2.2.1 to benefit from the most recent features and bug fixes (http://cufflinks.cbcb.umd.edu). [17:00:37] Loading reference annotation. [17:00:39] Inspecting reads and determining fragment length distribution. > Processed 31356 loci. [*************************] 100% > Map Properties: > Normalized Map Mass: 137260.00 > Raw Map Mass: 137260.00 > Fragment Length Distribution: Truncated Gaussian (default) > Default Mean: 200 > Default Std Dev: 80 [17:00:50] Estimating transcript abundances. > Processed 31356 loci. [*************************] 100% kitajima@kitajima[kitajima] |
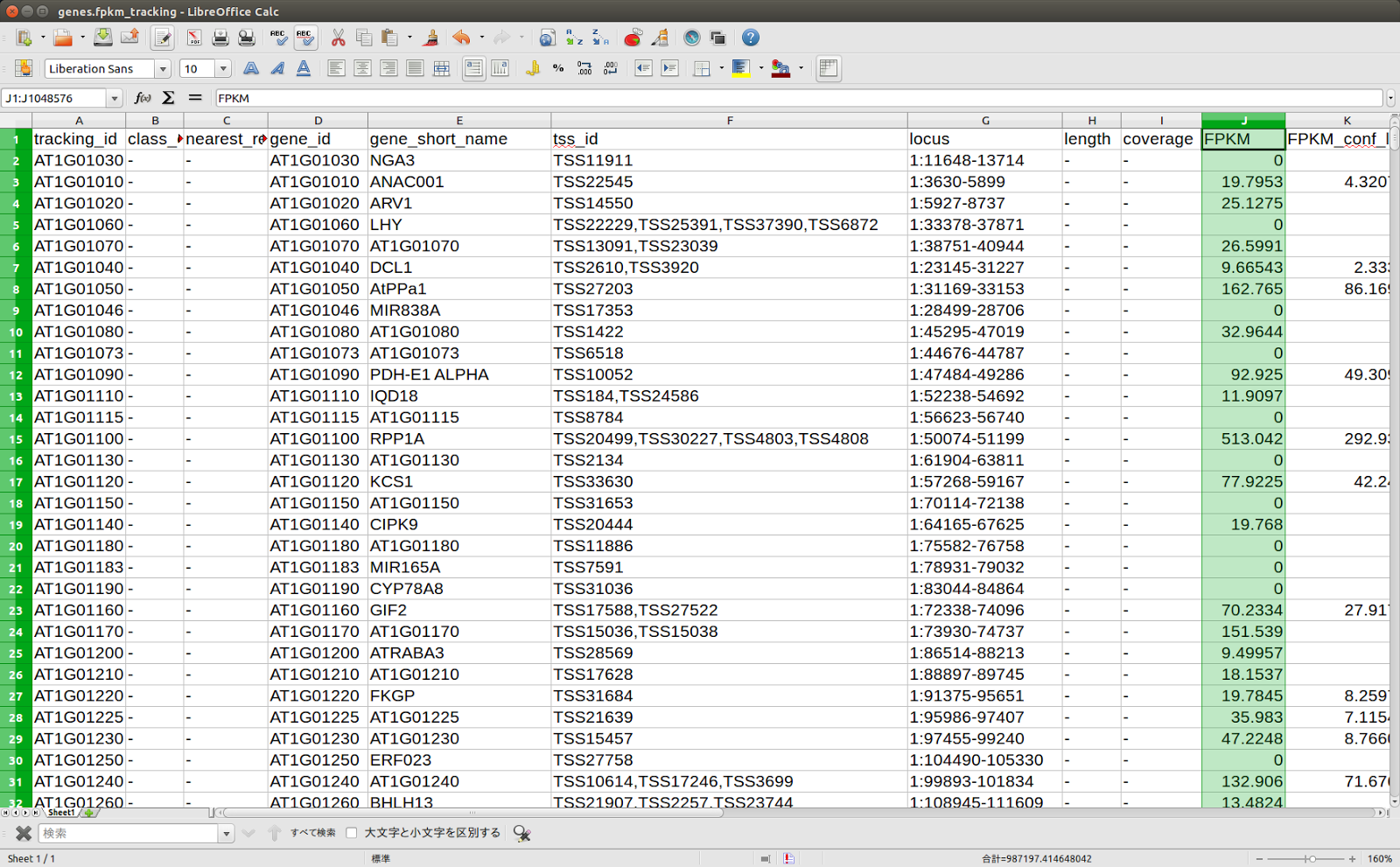
tophatresult_SRR401419ディレクトリにgenes.fpkm_trackingという名前のファイルができているはずだ。LibreOffice calcで開いてみよう。ファイルを読み込むときに区切りのオプションをきかれるはずなので、”タブ区切り”にチェックをいれ、他はオフにする。
アラビドプシスの場合、遺伝子にはATxGxxxxxというようなIDが割り当てられている。例えばAT1G01030は、Arabidopsis thalianaの第1染色体の第01030遺伝子、という具合だ。このファイルでは、第1および第4フィールドに遺伝子ID、第5フィールドに遺伝子の名称が並んでいる。第10フィールドにはFPKMという数値がかかれている。FPKMは、Fragments Per Kilobase of exon per Million mapped fragmentsの略で、テッポウユリのところで出てきたRPKMと似ている。RPKMがリファレンス配列にマップされたリードの数を表現する変数だったのに対し、FPKMでは、リードではなくcDNA断片の数を数えている。張り付いた断片の数を、RPKMの場合と同様、リファレンス配列1000塩基長当たりとリード総数100万当たりで標準化している。詳しくは、以下のブログを読むとよい。
http://shortreadbrothers.blogspot.jp/2011/06/fpkmrpkm.html
これで、発現ランキングができた。FPKM値の高い遺伝子は低い遺伝子よりも発現レベルが高い。
(3-4) シェルスクリプトで自動化してみる
(3-4-1) まずは書いてみる
同じようにして、のこり2つのsraファイルからFPKMランキング表を作ろう。ファイル名が異なるだけで作業内容自体は定型化しているので、シェルスクリプトにしておくと便利だ。以下の内容で、テキストファイルを作成し、~/work/sakitoにおいておく。ファイル名は、”sra2cufflinks_1.sh”としよう。見慣れない記号を解説しておくと、1行めの”#!/bin/bash”は、このファイルがシェルスクリプトであることを宣言している。2行め以降の#から始まる行は、コメント行なので、実行時には無視される。echo “------”と言う行は、”にはさまれた文字をモニタに表示せよ、というコマンドである。他の行は、すでにSRR401419で実施したコマンドと同じだ。ただし、ファイル名と出力先ディレクトリの名前だけは変えてある。
#!/bin/bash #本スクリプトの実行には、各SRRファイルが~/work/sakito/においてあることが前提。ただし、”sakito”の部分は各自のユーザー名に置き換える。以下も同様。 #Arabidopsisのgtfファイルは~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtfにあること。 #Tophat2で使用するbowtieは、暫定措置としてbowtie1を使う。 #Arabidopsisゲノム配列のbowtieindexは、~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/BowtieIndex/genomeにあること #-------------------------------- #sratoolkitによりsraをfastqに変換する。 echo “converting SRR401420.sra to fastq” ~/_Tools/sratoolkit/bin/fastq-dump ~/work/sakito/SRR401420.sra --split-files -O ~/work/sakito echo “converting SRR401421.sra to fastq” ~/_Tools/sratoolkit/bin/fastq-dump ~/work/sakito/SRR401421.sra --split-files -O ~/work/sakito #最初の120万行を抜き出してサブセットをつくる echo “creating subset of 1.2M lines” head -1200000 ~/work/sakito/SRR401420_1.fastq > ~/work/sakito/SRR401420_short_1.fastq head -1200000 ~/work/sakito/SRR401421_1.fastq > ~/work/sakito/SRR401421_short_1.fastq #Tophat2 with bowtie1 echo “Tophat2 with bowtie1 SRR401420_short_1” tophat2 --bowtie1 -p 3 -G ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf -o ~/work/sakito/tophatresult_SRR401420 ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/BowtieIndex/genome ~/work/sakito/SRR401420_short_1.fastq echo “Tophat2 with bowtie1 SRR401421_short_1” tophat2 --bowtie1 -p 3 -G ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf -o ~/work/sakito/tophatresult_SRR401421 ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/BowtieIndex/genome ~/work/sakito/SRR401421_short_1.fastq #samtoolsでソートする echo “samtools for sorting SRR401420_short_1” samtools sort ~/work/sakito/tophatresult_SRR401420/accepted_hits.bam ~/work/sakito/tophatresult_SRR401420/sort_accepted_hits echo “samtools for sorting SRR401421_short_1” samtools sort ~/work/sakito/tophatresult_SRR401421/accepted_hits.bam ~/work/sakito/tophatresult_SRR401421/sort_accepted_hits #cufflinksでFPKM計算 echo “cufflinks SRR401420_short_1” cufflinks -o ~/work/sakito/tophatresult_SRR401420/ -p 3 -G ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf ~/work/sakito/tophatresult_SRR401420/sort_accepted_hitst.bam echo “cufflinks SRR401421_short_1” cufflinks -o ~/work/sakito/tophatresult_SRR401421/ -p 3 -G ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf ~/work/sakito/tophatresult_SRR401421/sort_accepted_hits.bam |
sra2cufflinks_1.shファイルが出来上がったら、実行してみよう。以下のようにタイプしたら、終わるまで待っていればよい。しかし、エラーメッセージが出ていないか、ぐらいは確認したほうがよい。予期しない動作をしている場合は、controlを押しながらcとタイプすると強制終了できる。ともあれ、これで3回の繰り返し実験のFPKM値がそろったので、いつもの統計学解析に持ち込める。実験条件を変えて同じように測定を3回実施すれば統計学的な比較解析も可能だ。
kitajima@kitajima[kitajima] bash ~/work/sakito/sra2cufflinks_1.sh |
(3-4-2) もう少し高度なシェルスクリプト - 繰り返し処理 -
しかし、上のようなシェルスクリプトの書き方だと、同じような命令文を何度も書かなくてはならない。sraファイルの数が増えると、書き間違えそうだ。この様な時には、for文による繰り返し処理が有効だ。sra2cufflinks_1.shを以下のように手直ししてみる。sraファイルがいくつあろうとも、一連の処理は一回だけしか書いていない。その代わり、あらかじめどんなsraファイルがあるか教えて、それらについて同じ処理を繰り返すよう指示する。具体的には、スクリプト内の(1)では、SRR_arrayという配列を設定してSRR401419、SRR401420とSRR401421の3つの値を入力した。 (2)の行では、SRR_arrayに入っているSRR401419、SRR401420とSRR401421を、ひとつずつ順番にSRR_dataという変数に代入して、(3)のdoから(4)のdoneまでの間の処理を繰り返し実行しなさい、と指示している。 ${SRR_data}という文字列がところどころに使われているが、実行時には、ここにSRR401419、SRR401420とSRR401421が代入される。 このスクリプトをテキストファイルに保存して、~/work/sakitoディレクトリにおいておく。ファイル名は、”sra2cufflinks_2.sh”としよう。
#!/bin/bash #本スクリプトの実行には、各SRRファイルが~/work/sakito/においてあることが前提。ただし、”sakito”の部分は各自のユーザー名に置き換える。以下も同様。 #Arabidopsisのgtfファイルは~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtfにあること。 #Tophat2で使用するbowtieは、暫定措置としてbowtie1を使う。 #Arabidopsisゲノム配列のbowtieindexは、~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/BowtieIndex/genomeにあること #-------------------------------- # ↓(1) SRR_array=("SRR401419" "SRR401420" "SRR401421") # ↓(2) for SRR_data in "${SRR_array[@]}" # ↓(3) do echo "Analyzing $SRR_data" #sratoolkitによりsraをfastqに変換する。 echo “converting sra to fastq” ~/_Tools/sratoolkit/bin/fastq-dump ~/work/sakito/${SRR_data}.sra --split-files -O ~/work/sakito #最初の120万行を抜き出してサブセットをつくる echo “creating subset of 1.2M lines” head -1200000 ~/work/sakito/${SRR_data}_1.fastq > ~/work/sakito/${SRR_data}_short_1.fastq #Tophat2 with bowtie1 echo “Tophat2 with bowtie1” tophat2 --bowtie1 -p 3 -G ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf -o ~/work/sakito/tophatresult_${SRR_data} ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/BowtieIndex/genome ~/work/sakito/${SRR_data}_short_1.fastq #samtoolsでソートする echo “samtools for sorting” samtools sort ~/work/sakito/tophatresult_${SRR_data}/accepted_hits.bam ~/work/sakito/tophatresult_${SRR_data}/sort_accepted_hits #cufflinksでFPKM計算 echo “cufflinks” cufflinks -o ~/work/sakito/tophatresult_${SRR_data}/ -p 3 -G ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf ~/work/sakito/tophatresult_${SRR_data}/sort_accepted_hits.bam # ↓(4) done |
以下のようにタイプして実行する。
kitajima@kitajima[kitajima] bash ~/work/sakito/sra2cufflinks_2.sh |
(3-4-3) Rをつかって表を連結するシェルスクリプト
ここまでで、3つのsraファイルから各遺伝子のFPKM値を得た。tophatresult_SRR401419、tophatresult_SRR401420、tophatresult_SRR401421の各ディレクトリの中のgenes.fpkm_trackingというファイルにFPKM値が書き込まれている。これらをRで読み込んで手作業で連結することも可能であるが、sraファイルの数がもっと増えると大変なので、この作業もシェルスクリプトで自動化してみよう。次のシェルスクリプトでは、(1)の行で、echoコマンドをファイルへの書き込みに使っている。(1)から(2)までのスクリプトで、genes.fpkm_trackingから遺伝子IDとFPKMを抜き出して、sraファイル名_FPKM_list.csvというファイルに書き込んでいる。
また、(2)の”sed -e”というのは、指定する行を除去するコマンドである。(3)の”R --quiet --vanilla << EOF”の行から(4)のEOFと書いてある行までは、Rのスクリプトである。Rスクリプトの中では、ホームディレクトリから、ファイル名に”_FPKM_list.csv”という文字列を含むファイルをすべて読み込んで、遺伝子IDが揃うようにファイルをひとつずつ順番に連結している。最後に、結果をMerged_FPKM.csvに書きだす。このスクリプトをmerge.shと名づけて~/work/sakitoディレクトリに保存する。
#!/bin/bash #このスクリプトは、sra2cufflinks_2.shを実行した後につかう。 #-------------------------------- SRR_array=("SRR401419" "SRR401420" "SRR401421") #まず、遺伝子のID(第4フィールド)と遺伝子名(第5フィールド)を新しいファイルに書き出す。抽出元は変数$SRR_arrayの1番目に収納されているsraファイルの結果。 cat ~/work/sakito/tophatresult_${SRR_array}/genes.fpkm_tracking | cut -f4,5 > ~/work/sakito/Name_FPKM_list.csv for SRR_data in "${SRR_array[@]}" do #遺伝子IDとsraファイル名をタブ区切りで新しいファイルに書き込む。これがタイトル行になる。 # ↓(1) echo -e "gene_id""\t""${SRR_data}" > ~/work/sakito/${SRR_data}_FPKM_list.csv #それぞれのgenes.fpkm_trackingから第一行目(タイトル行)を削除した後、遺伝子ID(第4フィールド)とFPKM(第10フィールド)を抽出して追記する。 # ↓(2) cat ~/work/sakito/tophatresult_${SRR_data}/genes.fpkm_tracking |sed -e '1,1d'| cut -f4,10 >> ~/work/sakito/${SRR_data}_FPKM_list.csv done #------------------------------------------ #出力されたそれぞれのFPKMリストをマージして1つのcsvファイルにまとめる。 # Rにファイルを読み込ませるため作業ディレクトリをファイルのあるディレクトリに移行する。 cd ~/work/sakito # ↓(3) R --quiet --vanilla << EOF #以下、EOFまでRのスクリプト #~/work/sakitoディレクトリ内の_FPKM_list.csvファイルのファイル名を一括して変数fnamesに入力する。 fnames <- dir(path = "~/work/sakito", pattern="_FPKM_list.csv") fnames csvlist <- lapply(fnames,read.csv,header=T,sep="\t") #fnamesに格納したファイルの中身を順次csvlistに読み込まれる。ヘダーがカラム名であること、タブ区切りであることを指定。 names(csvlist) <- fnames #fnamesに格納したファイル名をnamesという変数にする。 n <- length(csvlist) n #ファイルの個数をnに入力。 # まず1つめのファイルの中身をtempに入力し、そこへ2つめ以降のファイルを順次マージする。all=Tは行が一致しない場合、足りないほうにNAを記入する。 temp <- csvlist[[1]] for (i in 2:n) { temp <- merge(temp, csvlist[[i]], all=T) } write.table(temp, file="~/work/sakito/Merged_FPKM.csv", row.names=FALSE, sep="\t") #マージの結果をMerged_FPKM.csvに書きだす。行番号を書かない、タブ区切りを指定。 # ↓(4) EOF #ホームディレクトリに戻る cd ~ |
以下のようにタイプして実行する。
kitajima@kitajima[kitajima] bash ~/work/sakito/merge.sh |
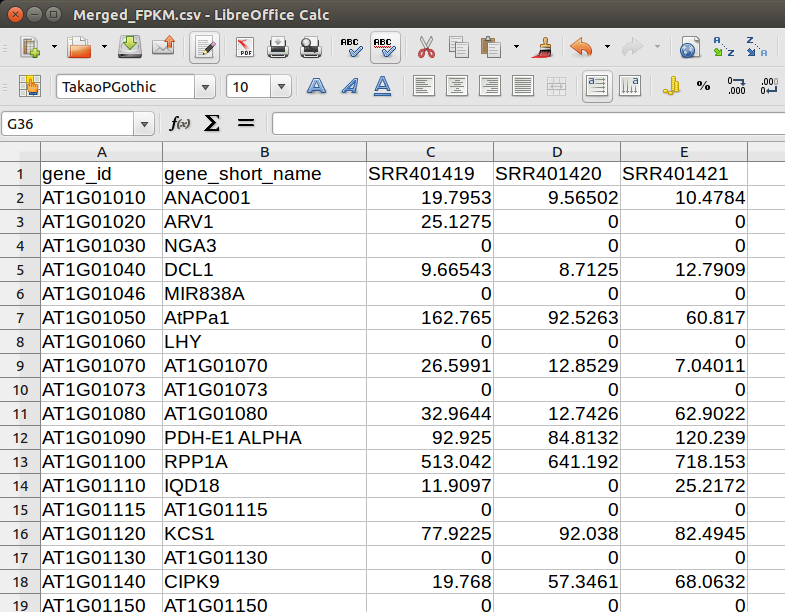
Merged_FPKM.csvをLibre Office calcで開くと右のようになっているはずだ。
(3-4-4) TopHat2の実行から結果ファイルの連結までを自動化するシェルスクリプト
ついでに、ここまで作ったsra2cufflinks_2.shとmerge.shをまとめて1つのシェルスクリプトにしておく。ファイル名は、sra2cufflinks_3.shとしておく。くりかえしになるが、もう一度書いておくと、このシェルスクリプトは、アラビドプシス用で、Bio-Linux 8での使用を前提にしている。TopHat2を実行するときに不本意ながらbowtie1を用いている。また、計算時間を短くするために、マッピングに使うfastqファイルは、オリジナルファイルの冒頭30万リードだけを抜き取ったものだ。解析したいsraファイルを指定するために、スクリプトの途中にある、”SRR_array=("SRR401419" "SRR401420" "SRR401421")”という行のカッコ内にsraファイル名を書く必要がある。実行すると、各sraファイルから計算されたFPKM値をMerged_FPKM.csvに書き出す。
#!/bin/bash #本スクリプトの実行には、各SRRファイルが~/work/sakito/においてあることが前提。ただし、”sakito”の部分は各自のユーザー名に置き換える。以下も同様。 #Arabidopsisのgtfファイルは~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtfにあること。 #Tophat2で使用するbowtieは、暫定措置としてbowtie1を使う。 #Arabidopsisゲノム配列のbowtieindexは、~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/BowtieIndex/genomeにあること #-------------------------------- #↓この行に解析したいsraファイル名を書く SRR_array=("SRR401419" "SRR401420" "SRR401421") for SRR_data in "${SRR_array[@]}" do echo "Analyzing $SRR_data" #sratoolkitによりsraをfastqに変換する。 echo “converting sra to fastq” ~/_Tools/sratoolkit/bin/fastq-dump ~/work/sakito/${SRR_data}.sra --split-files -O ~/work/sakito #最初の120万行を抜き出してサブセットをつくる echo “creating subset of 1.2M lines” head -1200000 ~/work/sakito/${SRR_data}_1.fastq > ~/work/sakito/${SRR_data}_short_1.fastq #Tophat2 with bowtie1 echo “Tophat2 with bowtie1” tophat2 --bowtie1 -p 3 -G ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf -o ~/work/sakito/tophatresult_${SRR_data} ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/BowtieIndex/genome ~/work/sakito/${SRR_data}_short_1.fastq #samtoolsでソートする echo “samtools for sorting” samtools sort ~/work/sakito/tophatresult_${SRR_data}/accepted_hits.bam ~/work/sakito/tophatresult_${SRR_data}/sort_accepted_hits #cufflinksでFPKM計算 echo “cufflinks” cufflinks -o ~/work/sakito/tophatresult_${SRR_data}/ -p 3 -G ~/work/sakito/Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf ~/work/sakito/tophatresult_${SRR_data}/sort_accepted_hits.bam #genes.fpkm_trackingから遺伝子IDとFPKM値を新しいファイルを作成する。 echo “遺伝子IDとFPKM値を抽出” #遺伝子IDとsraファイル名をタブ区切りで新しいファイルに書き込む。これがタイトル行になる。 echo -e "gene_id""\t""${SRR_data}" > ~/work/sakito/${SRR_data}_FPKM_list.csv #それぞれのgenes.fpkm_trackingから第一行目(タイトル行)を削除した後、遺伝子ID(第4フィールド)とFPKM(第10フィールド)を抽出して追記する。 cat ~/work/sakito/tophatresult_${SRR_data}/genes.fpkm_tracking |sed -e '1,1d'| cut -f4,10 >> ~/work/sakito/${SRR_data}_FPKM_list.csv done #genes.fpkm_trackingから遺伝子のID(第4フィールド)と遺伝子名(第5フィールド)を新しいファイルに書き出す。抽出元は変数$SRR_arrayの1番目に収納されているsraファイルの結果。 cat ~/work/sakito/tophatresult_${SRR_array}/genes.fpkm_tracking | cut -f4,5 > ~/work/sakito/Name_FPKM_list.csv #------------------------------------------ #出力されたそれぞれのFPKMリストをマージして1つのcsvファイルにまとめる。 # Rにファイルを読み込ませるため作業ディレクトリをファイルのあるディレクトリに移行する。 cd ~/work/sakito R --quiet --vanilla << EOF #以下、EOFまでRのスクリプト #~/work/sakitoディレクトリ内の_FPKM_list.csvファイルのファイル名を一括して変数fnamesに入力する。 fnames <- dir(path = "~/work/sakito", pattern="_FPKM_list.csv") fnames csvlist <- lapply(fnames,read.csv,header=T,sep="\t") #fnamesに格納したファイルの中身を順次csvlistに読み込まれる。ヘダーがカラム名であること、タブ区切りであることを指定。 names(csvlist) <- fnames #fnamesに格納したファイル名をnamesという変数にする。 n <- length(csvlist) n #ファイルの個数をnに入力。 # まず1つめのファイルの中身をtempに入力し、そこへ2つめ以降のファイルを順次マージする。all=Tは行が一致しない場合、足りないほうにNAを記入する。 temp <- csvlist[[1]] for (i in 2:n) { temp <- merge(temp, csvlist[[i]], all=T) } write.table(temp, file="~/work/sakito/Merged_FPKM.csv", row.names=FALSE, sep="\t") #マージの結果をMerged_FPKM.csvに書きだす。行番号を書かない、タブ区切りを指定。 EOF #ホームディレクトリに戻る cd ~ |
最後に、ここまで学んだことをどのようにして研究に活用するかを知ってもらうために、次の演習課題を挙げておく。
演習課題2 NCBI SRAデータベースに登録されているアクセッションNo.SRX117110 (http://www.ncbi.nlm.nih.gov/sra/SRX117110)に含まれる3つのsraデータ(SRR401428, SRR401429, SRR401430)は、ある変異体の花托のmRNA-seqデータで、上でもちいたSRR401419, SRR401420およびSRR401421はそれらの野性型コントロールである。これら6つのsraデータを使って、変異体と野性型の間での発現を比較し、統計学的有意さを考慮した上で大きな変動のある遺伝子を見付けなさい。もし可能なら、大きく発現変動した遺伝子がどのような機能をもつか調べて、この遺伝子変異が花の生理にどにのような影響を与えているのかを推察しなさい。 参考サイト; |
4. 参考文献その他 |
高速シーケンサーとそのデータ解析 ;
・ 菅野 純夫, 鈴木 穣, 次世代シークエンサー―目的別アドバンストメソッド 学研メディカル秀潤社 (2012/09) ISBN-10: 4780908558
・ 門田 幸二, トランスクリプトーム解析 (シリーズ Useful R 7), 共立出版 (2014/4) ISBN-10: 4320123700
シェルスクリプトの書き方 ;
・ 麻生二郎、誰でも簡単に書けるシェルスクリプト入門、日経Linux、第174号 (2014年3月号) p97-108
( http://mikke.g-search.jp/QNBP_LIN/2014/20140301/QNBP384876.html )
Linuxのコマンドと設定の全般 ;
コマンド自身にヘルプが埋め込まれているので、端末で、”コマンド --help”、”コマンド -help”、”コマンド -h”、あるいはコマンド名だけをタイプしてみるとよい。例えば、moreというコマンドをタイプすると、以下のように、どのようなオプションを指定できるか表示される。
kitajima@kitajima-BioLinux[kitajima] more Usage: more [options] file... Options: -d display help instead of ring bell -f count logical, rather than screen lines -l suppress pause after form feed -p suppress scroll, clean screen and disblay text -c suppress scroll, display text and clean line ends (以下省略) |
他にも、インターネット上に解説サイトや実施例が多数存在する。例えば、”Linux, sedの使い方”、”Linux, ファイルから特定の文字列を含む行を抽出する方法"、 “Linux, 2つのファイルを結合する方法"、"linux, 文字列を置換したい”、 というようなキーワードでググってみるとよい。 本実習で使用するBio-Linux 8の場合、バイオツール以外はUbuntu 14.04 LTSというLinuxそのものなので、OSレベルの一般的な設定方法を知りたい場合には、”Ubuntu, 日本語設定"というように検索するとよい。
各解析ツールの使用方法 ;
Linuxの一般的なコマンド同様、上記のような方法でヘルプを表示させることができる。また、そのツールのサイトを見ると詳細な英語での説明がある。多くの日本のユーザーが紹介ブログやトラブルシューティングを日本語で書いているので、それらも参考にするとよい。
補足資料
京都工芸繊維大学 情報科学センター Bio-Linux 8 利用の手引き
(2014-09-04)
● 初めに
この手引きでは、情報科学センターのシステムに固有なBio-Linux 8の使用方法について説明します。以下にある <ユーザ名> は情報科学センターアカウントのユーザ名です。
● NEBC Bio-Linux 8 標準インストールとの相違点
* ulimit -s unlimited されています
以下の追加ソフトウェアが ~/_Tools/ にインストールされています。端末でディレクトリに入り、ご利用ください。
* Trinity RNA-seq
* SRA Toolkit
* Velvet (MAXKMERLENGTH=99)
* Oases (MAXKMERLENGTH=99)
● NEBC Bio-Linux 8 の起動
1. 演習室の Windows に自身のユーザでログオンします。
2. すべてのプログラム→ Bio-Linux 8 を選択します。
3. VirtualBox のウィンドウが立ち上がり NEBC Bio-Linux が起動します。
● NEBC Bio-Linux 8 の終了
1. 画面左上端のアイコンをクリックし、メニューからシャットダウンを選びます。
2. ダイアログが出てきたらシャットダウンを選びます。
3. シャットダウン処理後、自動的に VirtualBox のウィンドウが閉じます(~/work/<ユーザー名>/のデータを除きすべてクリアされます)。
● 演習フォルダ
/home/manager/ (今回のシステムでは~/と同じ) は空き容量が少ないので、~/work/<ユーザー名>/ 配下で演習を実施してください。今回の実習で使用するシステムでは、まず端末で~/work/<ユーザー名>にカレントディレクトリを移すと、ユーザー名のディレクトリが自動的に作成されます。例えばアカウント名sakitoの場合、以下の青字部分のようにタイプします。
manager@bl8vbox[manager] cd work/sakito manager@bl8vbox[sakito] |
● 演習データの配布
演習データの配布にファイル共有サービスをご利用いただけます。
ウェブブラウザでhttps://proself01.cis.kit.ac.jp/にアクセスして、情報科学センターアカウントでログインします。
bioinfoフォルダが配布用の共有領域(注: 学生の情報科学センターアカウントでは読み取り専用です)
<ユーザ名>フォルダが個人領域(注: 情報科学センターアカウントの ~/Proself/ に紐付けられています。1GB 制限がありますのでご注意下さい)
になっています。
ファイル共有をディレクトリとして認識させたい場合は、ファイルマネージャー の”サーバーへ接続” を選んで
davs://<ユーザ名>@proself01.cis.kit.ac.jp/
に接続します。接続する際パスワードを求められますので、情報科学センターアカウントのパスワードを入力します。接続に成功すると、
/run/user/1001/gvfs/dav:host=fileshare.cis.kit.ac.jp,ssl=true,user=<ユーザ名>/
にマウントされます。