Abstract
-
Molecular dynamics simulation of helical structure formation in nanocylinder-confined single chains of isotactic polypropylene
-
Tomohiro Nakamura, Susumu Fujiwara, Katsumi Hagita
-
Helical structure formation in isotactic polypropylene (iPP) during cooling was investigated at the molecular level using the united-atom molecular dynamics simulations of single iPP chains confined in (i) repulsive-wall cylinders and (ii) carbon nanotubes (CNTs) with various chiral indices to investigate the effects of iPP–cylinder/ CNT interactions on iPP folding behavior. For both repulsive-wall cylinders and CNTs, the spreading length and number of folds depended on the tube diameter. The tube diameter was positively correlated with the number of folds and could therefore be adjusted to control the single-chain folding structure. Additionally, the single iPP chains did not adsorb to repulsive-wall cylinders but adsorbed to CNTs, which caused differences in folding behavior. These differences were ascribed to the corresponding spatial constraints: repulsive-wall cylinders provide a quasi-one-dimensional space that strongly constrains movement only in approximately one dimension, whereas CNTs have a two-dimensional curved surface. We believe that this study provides an important guiding principle for controlling the nanohybridization of iPP and CNTs.
-
Polymer 360, 130334 (2026).
-
-
PDBDNAConv: A software suite for generating atomistic DNA geometries for Geant4-DNA Monte Carlo simulations from Protein Data Bank (PDB) structures
-
Shun Fukagawa, Tsukasa Aso, Yuya Tanbo, Yoshiyuki Hirano, Susumu Fujiwara, Masanori Hara
-
Accurate simulation of radiation-induced DNA damage requires geometry models that preserve atomic structure while satisfying Geant4-DNA constraints. We present PDBDNAConv, a modular software suite that provides an automated pipeline for converting Protein Data Bank (PDB) structures into simulation-ready geometries. The software reconstructs hierarchical DNA models based on atomic structure using ellipsoidal approximation categorized for sugar, phosphate, and base moieties in nucleotide to reduce geometric complexes. Geometric overlaps are automatically detected and resolved to ensure valid geometry construction. Structured output is serialized in JSON format for interoperability and seamless integration into Geant4-DNA simulations. The exported JSON files are loadable by an example Geant4-DNA application through developed geometry construction classes that enable atomic-scale analysis of direct and indirect DNA damage.
-
SoftwareX 34, 102746 (2026).
-
-
Accessibility of deoxyribose hydrogens to hydroxyl radicals in nucleosomal and naked DNA in water: a molecular dynamics study with LAMMPS
-
Yoshiyuki Hirano, Kanta Nambu, Tsukasa Aso, Masanori Hara and Susumu Fujiwara
-
Ionizing radiation induces DNA damage both directly and indirectly through hydroxyl radicals (·OH) generated by water radiolysis. The indirect action involves hydrogen abstraction from deoxyribose, leading to DNA damage such as base release and strand breaks. The probability of such events depends on the accessibility of specific hydrogen atoms to ·OH radicals. Previous computational studies have examined naked DNA fragments; however, the effect of nucleosomal organzation has not been addressed. Here, we performed molecular dynamics (MD) simulations using LAMMPS (Large-scale Atomic Molecular Massively Parallel Simulator) to evaluate the accessibility of ·OH radicals to deoxyribose hydrogens in both 12-bp naked B-type DNA and nucleosomal DNA containing histone proteins. The accessibility to the hydrogens increased, following the order H1'≈H2'≈H3'<H4'<H5', consistent with previous studies. Accessibility was approximately proportional to the solvent accessible surface area (SASA), supporting SASA as a practical predictor of accessibility even in base-dependent differences were observed. In nucleosomal DNA, accessibility of hydrogens facing the histone core was significantly reduced compared to solvent-facing hydrogens, despite comparable SASA values. This reduction suggests that histones physically hinder ·OH radical penetration. These findings highlight the importance of considering protein−DNA complexes when modeling indirect radiation effects.
-
Phys. Scr. 101, 035001 (2026).
-
-
Reactive molecular dynamics simulations of the intra- and intermolecular reactions of hydrogen-abstracted polyethylene chains
-
Haolun Li, Susumu Fujiwara, Hiroaki Nakamura, Tomoko Mizuguchi, Shinji Saito, Wataru Sakai
-
Polymers can be radicalized by radiation, and the reaction of the generated radicals with each other could result in structural changes in the original material. Our previous reactive molecular dynamics (MD) simulations showed that several types of reactions occur at specific locations in polymer structures at low temperatures. However, not much is known about the occurrence of other types of reactions for these materials. In this study, reactive MD simulations were performed on hydrogen-abstracted polyethylene chains to clarify the conditions under which other types of reactions, such as chain scission and crosslinking, occur. Our simulation results showed that chain scission occurred only at a specific location at temperatures of 348 K or higher. Intermolecular crosslinking occurred only when two CH radicals were fixed at a distance of approximately 2.0 Å apart. However, our simulations also indicated that such movement does not occur freely owing to repulsive forces. These findings provide a new understanding of the structural changes and chemical reactions of polymers caused by radiation damage.
-
Mol. Simul. 51(2), 122-127 (2025).
-
-
Molecular dynamics study on clustered DNA damage: AP sites on the same strand
-
Kazushi Terakawa, Susumu Fujiwara, Tomoko Mizuguchi, Hiroaki Nakamura, Ken Akamatsu, Naoya Shikazono, Yoshiteru Yonetani
-
Although radiation-induced clustered DNA damage can have critical biological consequences, the underlying molecular mechanisms remain unclear. To explore the effect of clustered DNA damage on DNA structure and dynamics, we performed molecular dynamics simulations on damaged DNA with two AP sites on the same strand, that is, a tandem AP cluster. The results showed that the cluster insertion of the two AP sites had a significant impact on the DNA's local and global structures. Local structural deformations as well as the extrahelical form, AP-base pairs, and irregular base pairs were frequently observed. Unlike a single AP site, the tandem AP cluster revealed that these local structural features occurred simultaneously within a small separation. Moreover, we found that the presence of tandem AP sites induced global bending of DNA. This suggests that the present case with tandem AP sites may have a non-negligible impact on the biological function of damage repair.
-
Biophys. Chem. 319, 107394 (2025).
-
-
Reactive molecular dynamics simulation on DNA double-strand breaks induced by
hydrogen elimination
-
Hiroaki Nakamura, Kento Ishiguro, Ayako Nakata, Shunsuke Usami, Seiki Saito, and Susumu Fujiwara
-
We propose a scar model to simulate double-strand breaks (DSBs) in telomeric DNA induced by the β-decay of tritium into helium. The two hydrogens bonded to the 5' carbon connecting the pentasaccharides and phosphate are removed in this scar model. Molecular dynamics simulations using a reactive force field were conducted across ten cases involving telomeric DNA composed of 16 base pairs (32 nucleotides). DSBs are observed in structures with more than 24 scars, whereas only single-strand breaks (SSBs) occur in the case with 16 scars. Moreover, in the case of {16,0} and {0,16}, where only one of the strands had scars, SSB occurs only in the scarred strand. Second, in the {16,8} and {8, 16} cases, DSBs occur. Thus, we conclude that the following conditions are necessary for DSBs: (i) scars must be present on both the left (L) and right (R) strands and (ii) a substantial number of scars (24 or more) must be in close proximity.
-
Jpn. J. Appl. Phys. 63, 12SP09 (2024).
-
-
Analyzing hydroxyl radical accessibility related to hydrogen abstraction in a DNA sugar moiety using molecular dynamics simulations
-
Ryuta Kawanami, Susumu Fujiwara, Yoshiteru Yonetani, and Tsukasa Aso
-
Exposure of water to ionizing radiation induces OH radical formation. Within cellular environments, the presence of OH radicals can stimulate the abstraction of hydrogen atoms from the sugar backbone of DNA. Subsequent damage to DNA structures leads to various diseases. Multiple studies have elucidated this phenomenon, especially computational studies examining the differences in the degree of abstraction between the sugar hydrogens (H1', H2', H2'', H3', H4', H5', and H5''). However, the details of this phenomenon have not yet been clarified. It is rare for OH radicals to approach DNA within the simulation time, making it difficult to adequately sample the configurations in which OH radicals immediately precede the abstraction of the hydrogen atoms. To address this problem, we performed molecular dynamics simulation to calculate the relative accessibility by putting a potential on nucleotides and OH radicals. As a result, we found that the accessibility of OH radicals to each hydrogen atom differs from that of water molecules as solvents. A more detailed accessibility analysis revealed that the angle of the OH radicals approaching the hydrogen atoms of ribose and the energy barrier for abstracting the hydrogen atoms can be considered to improve the correspondence with the experimental data. Moreover, we found that the behavior of water molecules and OH radicals toward accessibility to DNA differs significantly and showed that the factors are related to the physicochemical properties of water molecules and OH radicals, as well as the structure of DNA.
-
AIP Advances 14, 105318 (2024).
-
-
Molecular dynamics study of perturbation on protocell membrane induced by magnesium ion
-
Ryuta Kawanami and Susumu Fujiwara
-
Among research of origin of life, vesicles are often studied as protocells. Fatty acids are synthesized abiotically, and their properties have been investigated as one of the most promising candidates for protocell membranes.
Magnesium ions Mg2+ in particular are thought to have played an important role for ribozymes.
However, it has been found that in the presence of magnesium ions, protocells of pure fatty acids cannot exist or retain its contents.
In this study, we analyzed the response of fatty acid bilayers to Mg2+ using molecular dynamics simulations to elucidate the mechanism of vesicle destabilization.
In the results, tighter packing of fatty acid molecules was shown.
It was also found that adding Mg2+ causes attraction between ionized fatty acid molecules.
These results suggest that the destabilization of fatty acid vesicles by Mg2+ may be due to pore formation by shrinkage of the outer leaflet of the vesicles.
-
J. Adv. Simulat. Sci. Eng. 11(1), 179-187 (2024).
-
-
Molecular Dynamics of Topological Barriers on the Crystallization Behavior of Ring Polyethylene Melts with Trefoil Knots
-
Katsumi Hagita, Takahiro Murashima, Naoki Sakata, Koya Shimokawa, Tetsuo Deguchi, Erica Uehara, and Susumu Fujiwara
-
Topological barriers in ring polyethylene (PE) melts of trefoil knots inhibit crystallization due to self-entanglement, as confirmed by united atom molecular dynamics (UAMD) simulations. In this study, we clarified the decrease in the topological barriers of self-entangled knots with increasing polymer chain length (N) and the localization of the knotted segments in the noncrystallized region. In the UAMD simulations, isothermal crystallization processes were performed at T = 300 K for the trefoil knots using a crystallization time tIC = 200 ns. Here, the trefoil knot gives the simplest nontrivial topology with nonzero crossing number. Crystallization was not observed in trefoil PE knots for N ≤ 120; however, crystallization did take place in the ring PE melt of the trivial knot with trivial topology and zero minimal crossing number. For N = 140, suppression of the growth of a crystalline phase (i.e., the polycrystalline phase) was observed in the trefoil PE melts, whereas such suppression was not detected in the trivial PE melts. In contrast, no significant differences in the crystallization behaviors of the trefoil and trivial PE melts were observed at N = 200. These results indicated that the topological barriers decreased with increasing N. Furthermore, to investigate the relationship between the chain conformation and degree of local crystallization, we developed a new mathematical method for searching the knotted segment of a trefoil knot in a crystallized trefoil PE melt. We found that trefoil knots with large subloops (“large-leaves”) exhibited localization of the knotted segment. In addition, the large leaves of the localized knots dominated in the crystallized region, and the knotted segments of the localized knots were located mainly in the noncrystallized region.
-
Macromolecules 56, 15-27 (2023).
-
-
Linear response theory-based theoretical approach to structural changes in a polymer induced by β-decay of substituted tritium
-
Ryuta Kawanami, Susumu Fujiwara, Hiroaki Nakamura and Kazumi Omata
-
Polymers exposed to tritiated water undergo hydrogen defects caused by isotope substitution and subsequent β-decay of substituted tritium, causing structural changes and loss of function in the biopolymers. Here, based on linear response theory, we predict the structural change of tritium-damaged polyethylene using the equilibrium trajectory of undamaged polyethylene to reduce the computation time of molecular dynamics simulations. Specifically, the ensemble average of the change in a physical quantity, such that it represents a structural change before and after damage, was calculated numerically using the time derivative of the total potential energy difference derived analytically and the physical quantity obtained from the simulation of undamaged polyethylene on the basis of linear response theory. A comparison between theoretical and simulation results revealed that the characteristic oscillation behaviors of the structural response of polyethylene can be predicted, whereas the quantitative prediction of the steady-state values over a long period is difficult.
-
Jpn. J. Appl. Phys. 62, SA1001 (2023).
-
-
Rate of double strand breaks of genome-sized DNA in tritiated water: Its dependence on tritium concentration and water temperature
-
Yuji Hatano, Hiroto Shimoyachi, Tatsuya Asano, Takahiro Kenmotsu, Takuro Wada, Yasuhisa Oya, Hiroaki Nakamura, Susumu Fujiwara
-
The goal of this study is to establish a simple experimental system to examine the rate of double strand breaks (DSBs) of genome-sized DNA molecules under irradiation of β-rays from tritium under well-controlled conditions for the validation of computer simulation on interactions of biomolecules and ionizing radiation. Irradiation effects were insignificant at tritium concentration of 1300 Bq/cm3, indicating that the effects of β-rays were far smaller than those of oxidation and/or thermal motion at the low dose rate (4.3 μGy/h). Clear increase in DSB rate was observed at tritium concentrations of 3.0-4.0 MBq/cm3. The temperature de-pendence of DSB rate was examined by using the high concentration tritiated water.
-
J. Adv. Simulat. Sci. Eng. 9(1), 198-205 (2022).
-
-
The study on the stability of DNA structure by steered molecular dynamics simul\
ations
-
Tomoko Mizuguchi, Naoto Fukushima, Takashi Aoki, Susumu Fujiwara, Masato Hashimoto
-
The stability of DNA double-stranded structure is examined by pulling atoms using steered molecular dynamics simulations. We use the base sequence to which DNA helicase binds; it acts to separate a double-stranded chain into single-stranded ones. The force is applied to atoms in the middle of the strand and they are pulled perpendicular to the helical axis of DNA. The force profile basically corresponds to the fraction of Watson-Crick hydrogen bonds which shows jumps and plateaus. The work for double-stranded separation can be interpreted in conjunction with the way to break hydrogen bonds. When some hydrogen bonds are broken at once, the bigger force is needed, and it leads to morework.
-
J. Adv. Simulat. Sci. Eng. 9(1), 160-169 (2022).
-
-
A theoretical approach to structural change of a polymer induced by beta decays of substituted tritium based on the linear response theory
-
Susumu Fujiwara, Ryuta Kawanami, Haolun Li, Hiroaki Nakamura, Kazumi Omata
-
Molecular dynamics simulations of the hydrogen-removed polyethylene are carriedout to study the structural change of polyethylene induced by beta decays of substituted tritium. Our simulations show that the folded structure of the hydrogen-removed polyethylene becomes more disordered as the number of removed hydrogen atoms becomes larger. We also propose a theoretical approach to explaining and predicting our molecular dynamics simulation results of hydrogen-removed polyethylene on the basis of the linear response theory. We derive the time derivative of the dynamical quantity, which is conjugate to the force applied as perturbation in the framework of the linear response theory, required to calculate the response function. The dynamical quantity in this study is the total potential energy difference of polyethylene before and after removal of hydrogen. Preliminary results of the response function for the total potential energy of polyethylene after removal of hydrogen are presented.
-
J. Adv. Simulat. Sci. Eng. 8(2), 211-222 (2021).
-
-
Image processing method for automatic measurement of number of DNA breaks
-
Seiki Saito, Hiroaki Nakamura, Takahiro Kenmotsu, Yasuhisa Oya, Yuji Hatano, Yuichi Tamura, Susumu Fujiwara and Hiroaki Ohtani
-
The number of double-strand breaks can be evaluated from the change of average DNA length. The average DNA length is measured by the single-molecule observation method using fluorescence microscope. The measurement of DNA length in the microscope images is done manually by experienced operators and it is time consuming in many experiments. An image processing method using OpenCV library to measure length of DNA in fluorescence microscope images is developed in this paper. An automation of measurement using deep learning is also proposed.
-
J. Adv. Simulat. Sci. Eng. 8(2), 173-193 (2021).
-
-
Evaluation of Solvent-Accessible Surface Area of Backbone Hydrogen in Telomeric DNA Using Molecular Dynamics Simulation Molecular dynamics simulations for structure formation of polymers
-
Yohei Tsuchida, Seiki Saito, Hiroaki Nakamura, Yoshiteru Yonetani, Susumu Fujiwara
-
Tritiated water is generated under the decommissioning process of the Fukushima Daiichi Nuclear Power Station. The Ministry of Economy, Trade and Industry, Japan (METI) and Tokyo Electric Power Company Holdings (TEPCO) are considering releasing tritium into the ocean. In addition, tritium is planning to use as fuel in fusion power plants, which is expected as a future power generation technology. Therefore, it is important to understand the impact of tritium on biomolecules in living organisms including human in detail. We aim to elucidate the mechanism of DNA damage due to the radioactive decay effect that occurs when light hydrogen in human DNA is replaced with tritium, using molecular dynamics (MD) methods. To understand the decay effect on DNA, first, it is necessary to evaluate the replaceability of light hydrogen to tritium for each hydrogen in DNA. In this study, to evaluate the degree of replaceability of the backbone hydrogen atoms in telomeric DNA of human, solvent-accessible surface area (SASA) is calculated for the data obtained by MD simulation. As a result, it is found that the SASA of H5 hydrogen is large in the hydrogen atoms in the backbone.
-
Trans. Jpn. Soc. Simul. Tech. 13(1), 32-36 (2021) [in Japanese].
-
-
Structural change of damaged polyethylene by beta-decay of substituted tritium
using reactive force field
-
Haolun Li, Susumu Fujiwara, Hiroaki Nakamura, Tomoko Mizuguchi, Ayako Nakata, Tsuyoshi Miyazaki and Shinji Saito
-
The molecular mechanism of structural change caused by the beta-decay of substituted tritium on DNA or polymeric materials is still being unsolved and it is hard to study the decay effect of tritium solely by experiment. In order to study the structural changes of damaged polyethylene caused by the decay effect of tritium, we randomly removed hydrogen atoms from the polyethylene chain and performed molecular dynamics (MD) simulations using the reactive force field (ReaxFF). We adopted two parameter sets of ReaxFF and evaluated their reliability by comparing the atomic forces with density functional theory calculations. The results of MD simulations at a low temperature of 100 K show that the structure of polyethylene will be less ordered when losing more hydrogen atoms. It is observed that a double bond or a cyclic structure will be formed when two carbon atoms, which are the nearest or next-nearest neighbors, lose hydrogen atoms.
-
Jpn. J. Appl. Phys. 60, SAAB06 (2021).
-
-
Icosahedral order in liquid and glassy phases of cyclohexane
-
Tomoko Mizuguchi, Soichi Tatsumi and Susumu Fujiwara
-
We performed all-atom molecular dynamics simulations for bulk cyclohexane and
analysed the short- and medium-range structures in supercooled and glassy
states by using the Voronoi tessellation technique.
From the analyses of both the potential energy of the system and the radial
distribution function of molecules, cyclohexane was found to be vitrified as
the temperature decreased. Furthermore, the icosahedral-like local structures
are dominant at all temperatures and grow in a supercooled liquid, whereas
the face-centred cubic structures do not grow when the temperature decreases.
It was also ascertained that the icosahedral-like structure is more dominant
than the full-icosahedral one.
The network of the distorted icosahedron spreads throughout the system at
low temperatures.
Our simulation demonstrates the stability of the icosahedral local structure
even in a non-spherical molecule such as cyclohexane.
-
Mol. Simul. 46(10), 721-726 (2020).
-
-
Molecular dynamics study on DNA damage by tritium disintegration
-
Hiroaki Nakamura, Hisanori Miyanishi, Takuo Yasunaga, Susumu Fujiwara,
Tomoko Mizuguchi, Ayako Nakata, Tsuyoshi Miyazaki, Takao Otsuka,
Takahiro Kenmotsu, Yuji Hatano and Shinji Saito
-
Using molecular dynamics (MD) simulation, we simulate the structural change
of a telomeric DNA by β-decay of substituted
tritium to helium-3.
he configuration of the telomeric DNA is obtained by removing TRF2 protein
from the TRF2-Dbd-DNA complex (Protein Data Bank ID is 3SJM).
We assume that hydrogens (H) of guanines in the telomeric DNA are replaced
to helium-3. Since this replacement of the H atoms to the 3He atoms changes
the charge distribution significantly, the charge distribution used
in the MD simulation for the modified guanine is obtained by the density
functional theory calculations. We adopt, as the MD simulation,
nanoscale molecular dynamics code with CHARMM36 force field using Langevin
thermostat and Nosé-Hoover Langevin piston
to control the temperature and pressure of the system, respectively.
Moreover, changing both the number
of replaced guanine N and the temperature of the system T, we calculate
the root mean square deviation RMSD to quantify the dependence of the
durability of the telomeric DNA on the β-decays.
From the MD simulation, it is found that as N or T becomes larger,
the RMSD of the DNA becomes also larger.
Namely, it denotes that as the intensity of the
β-decays becomes arger or as the temperature
is increased, the DNA structure becomes more fragile.
-
Jpn. J. Appl. Phys. 59, SAAE01 (2020).
-
-
Single-chain folding of a quenched isotactic polypropylene chain through united
atom molecular dynamics simulations
-
Katsumi Hagita, Susumu Fujiwara
-
The single-chain folding of an isotactic polypropylene (iPP) chain through
united atom (UA) molecular dynamics simulations under quenching was
investigated using force fields (FFs) based on TraPPE-UA.
We estimated the degree of local rigidity of the folded chain along iPP
undergoing single-chain folding. To maintain the tacticity of iPP,
we introduced improper torsional angle potentials and/or explicit hydrogen
atoms bonded to backbone carbon atoms. In the simulation using modified
TraPPE-UA FFs with added hydrogen atoms, folded structures were observed.
In the cases using modified TraPPE-UA FFs with only improper torsional
potentials, folding of the quenched iPP chain was not observed.
Moreover, to clarify the folding behavior of the iPP chain, we studied
the chirality of a single iPP chain and subsequently observed spontaneous
chirality selection during the quenched folding process.
We also examined the effect of the angle and torsional potentials of
the added hydrogen on the folding behavior of a single iPP chain into
local crystals. Therefore, we confirmed that the strength of the angle
and torsional potentials contributed to the acceleration of the folding
behavior.
-
Polymer 183, 121861 (2019).
-
-
Hydrogen bond analysis of confined water in mesoporous silica using the reactive force field
-
Tomoko Mizuguchi, Katsumi Hagita, Susumu Fujiwara and Takeshi Yamada
-
The structural and dynamical properties of water confined in nanoporous
silica with a pore diameter of 2.7 nm were investigated by performing
large-scale molecular dynamics simulations using the reactive force field.
The radial distribution function and diffusion coefficient of water were
calculated, and the values at the centre of the pore agreed well with
experimental values for real water. In addition, the pore was divided
into thin coaxial layers, and the average number of hydrogen bonds,
hydrogen bond lifetime and hydrogen bond strength were calculated as
a function of the radial distance from the pore central axis.
The analysis showed that hydrogen bonds involving silanol (Si-OH) have
a longer lifetime, although the average number of hydrogen bonds per atom
does not change from that at the pore centre. The longer lifetime,
as well as smaller diffusion coefficient, of these hydrogen bonds is
attributed to their greater strength.
-
Mol. Simul. 45, 1437-1446 (2019).
-
-
Structural change of tritium-substituted polymeric materials by a beta decay:
A molecular dynamics study
-
Haolun Li, Susumu Fujiwara, Hiroaki Nakamura, Tomoko Mizuguchi, Takuo Yasunaga,\
Takao Otsuka, Takahiro Kenmotsu, Yuji Hatano, Shinji Saito
-
The molecular mechanism through which how beta decays in tritium-substituted
species damage DNA and polymeric materials is still unknown.
Molecular dynamics simulations of hydrogen-removed polyethylene were
performed to predict the structural change of the polyethylene chain
after the substituted tritium decays. We calculated the potential energy,
the global orientational order parameter, and the average number of
consecutive trans bonds. The results are that, the greater the number of
removed hydrogen atoms, the higher the potential energy and the lower
the value of the global orientational order parameter and the average
number of consecutive trans bonds. Thus, after losing hydrogen,
polyethylene becomes poorer in terms of both thermal and structural
stabilities.
-
Plasma Fusion Res. 14, 3401106 (2019).
-
-
Volume Rendering Method Applied to 3D Edge Impurity Emission in LHD to Produce
Projection Image in Arbitrary Plane
-
Yuichi Tamura, Masahiro Kobayashi, Taisuke Kobayashi, Wataru Omori, Hiroaki Nakamura, Hiroaki Ohtani, Susumu Fujiwara and the LHD Experimental Group
-
Understanding edge impurity transport is one of the important issues for
fusion devices to control edge radiation distribution for detachment
operation and impurity influx to the confinement region.
In LHD, the edge magnetic field structure becomes complex stochastic magnetic
field. In order to study relation between impurity transport and the magnetic
field geometry, 3D edge impurity emission distributions are obtained by
a multichannel spectrometer system and tomography scheme.
However, it is difficult to understand the three-dimensional (3-D) structure.
Therefore, we propose a visualization system that employs a volume rendering
method. With the proposed system, which can be used on a PC or mobile device,
the user can observe a 3D structure in an arbitrary plane.
To realize this function, we propose a volume visualization system
comprising preprocessing and real-time rendering stages.
Therefore, the visualization framerate can exceed 30 frames per second on
PCs and approximately six frames per second on mobile devices,
although the user frequently changes the position and direction of the camera.
-
Plasma Fusion Res. 14, 3401106 (2019).
-
-
Computational strategy for studying structural change of tritium-substituted macromolecules by a beta decay to helium-3
-
Susumu Fujiwara, Hiroaki Nakamura, Haolun Li, Hisanori Miyanishi, Tomoko Mizuguchi, Takuo Yasunaga, Takao Otsuka, Yuji Hatano, Shinji Saito
-
We propose a computational strategy for investigating structural change of
tritium-substituted macromolecules. Effects of radiation on macromolecules
such as polymeric materials and DNA are classified into three categories:
(1) direct action, (2) indirect action, and (3) decay effect. In this study,
we focus on the decay effect exclusively. After a beta decay of substituted
tritium in macromolecules to helium-3, the generated inert helium-3 is assumed
to be deleted quickly. To get an insight into the decay effect to the damage
of macromolecules, we perform molecular dynamics simulations of
tritium-deleted macromolecules and analyze their structural change.
Preliminary simulation results of decay effect on polymeric materials and
DNA are presented.
-
J. Adv. Simulat. Sci. Eng. 6(1), 94-99 (2019).
-
-
Cavitation in Thin Films of Amorphous Polymers from the Static Melt Induced by T
hermal Treatment
-
Masato Hashimoto, Susumu Fujiwara, Nan Lin, Atsushi Doi,
Nozomi Katayama, Junki Ootani and Tomoko Mizuguchi
-
We discover a new cavitation phenomenon in amorphous polymers sandwiched
between two thick slide glasses from the static melt induced by thermal
treatment. By quenching atactic polystyrene (aPS) samples from the static
melt under the glass transition temperature (Tg) and annealing
them above Tg for thick slide glasses, cavities are created to relax the
negative pressure. Cavity growth undergoes an Ostwald ripening-like process
in cases of large molecular weight, while it undergoes a viscous fingering
process in cases of small molecular weight. The induction time for cavity
formation is found to decrease with the increase of the annealing
temperature and with the decrease of the molecular weight. By contrast,
no cavities are observed in an aPS sample between two thin cover glasses
because the negative pressure can be relaxed by bending the whole thin
cover glasses.
-
Polymer J. 51, 569-577 (2019).
-
-
An Accelerated United-Atom Molecular Dynamics Simulation on the Fast Crystallization of Ring Polyethylene Melts
-
Katsumi Hagita, Susumu Fujiwara, and Nobuyuki Iwaoka
-
To investigate crystallinities based on trans structures, we determined the
differences in the crystallization properties of ring and linear polymers by
performing united-atom-model molecular dynamics (MD) simulations of
homogenous polyethylene melts of equal length, N, which refers to the number of
monomers per chain. Modified parameters based on the DREIDING force field (FF)
for the CH2units were used in order to accelerate the
crystallization process. To detect polymer crystallization, we introduced
some local-order parameters that relate to trans segments in addition to
common crystallinities using neighboring bond orders. Through quenching MD
simulations at 5 K/ns, we roughly determined temperature thresholds,
Tth, at which crystallization is observed, although it was hard
to determine the precise Tth as observed in the laboratory time
frame with the present computing resources. When N was relatively small
(100 and 200), Tth was determined to be 320 and 350 K for the
linear- and ring-polyethylene melts, respectively, while Tth
was found to be 330 and 350 K, respectively, when N was 1000. Having
confirmed that the crystallization of a ring-polyethylene melt occurs
faster than that of the analogous linear melt, we conclude that the
trans-segment-based crystallinities are effective for the analysis of
local crystal behavior.
-
J. Chem. Phys. 150, 074901 (2019).
-
-
Structure formation of a quenched single polyethylene chain with different force fields in united atom molecular dynamics simulations
-
Katsumi Hagita, Susumu Fujiwara and Nobuyuki Iwaoka
-
The differences in the structure formations of a single polyethylene (PE)
chain in united atom molecular dynamics (UAMD) simulations under quenching
was investigated with varying force fields (FFs) that included torsional
potential.
We estimated the crystallinity of the folded structures undergoing local
crystallization of the single PE chain.
In simulations with DREIDING FF, highly folded structures were observed
with fast quenching at the rate of 50 K/ns.
From the viewpoint of crystallinity, we clarified that it was easy to
achieve folding of the PE chains into local crystals with DREIDING FFs.
With recent commonly used general FFs such as OPLS-UA and TraPPE-UA, highly
folded structures were observed by quenching at the rate of 1 K/5 ns.
In the present paper, we examine the Rigby-Roe (RR) FF optimized by
Theodorou and coworkers and Paul-Yoon-Smith (PYS) FF optimized by Rutledge
and coworkers.
With the RR-Theodorou and PYS-Rutledge FFs, crystallization was observed
with quenching at 1 K/ns and 1 K/5 ns, respectively.
Consequently, it was relatively easier to achieve folding with the
RR-Theodorou FF than with the PYS-Rutledge, OPLS-UA, and TraPPE-UA FFs.
-
AIP Advances 8(11), 115108 (2018).
-
-
Dissipative Particle Dynamics Simulation for Self-Assembly of Symmetric Bolaamphiphilic Molecules in Solution
-
Susumu Fujiwara, Yoshiki Iida, Takehide Tsutsui, Tomoko Mizuguchi, Masato Hashimoto, Yuichi Tamura and Hiroaki Nakamura
-
The self-assembly of dissolved symmetric bolaamphiphilic molecules is studied
using dissipative particle dynamics simulations.
Specifically, we investigate how interactions between the dual hydrophilic
ends of the molecules affect the self-assembly process.
Simulations show that four types of self-assembled structures (spherical
micelles, tubes, vesicles, and wormlike micelles) are obtained from
a random configuration of symmetric bolaamphiphilic molecules in solution.
We find that the self-assembled structures change from spherical micelles
to tubes, then to vesicles, and finally to wormlike micelles as the repulsive
interactions between the hydrophilic ends increase.
The molecular shapes in vesicles tend to be more rodlike than those in
spherical micelles, tubes, or wormlike micelles.
-
Plasma Fusion Res. 13, 3401095 (2018).
-
-
Dissipative Particle Dynamics Simulation of Self-Assembly in a Bolaamphiphilic Solution
-
Susumu Fujiwara, Yu Takahashi, Hiroki Ikebe, Tomoko Mizuguchi, Masato Hashimoto,
Yuichi Tamura, Hiroaki Nakamura and Ritoku Horiuchi
-
The self-assembly of flexible bolaamphiphilic molecules in a solution is
studied by dissipative particle dynamics simulations. In particular,
we investigate the effect of the interaction difference, Δa, between
the two different hydrophilic end groups on the self-assembly in a
bolaamphiphilic solution. Our simulations show that two types of
self-assembled structures, spherical vesicles and worm-like micelles, are
obtained from a random configuration of bolaamphiphilic molecules in a solution.
We find that the worm-like micelles are formed when Δa > 0,
whereas spherical vesicles are obtained when Δa ≤ 0. It is also
ascertained that the size of the spherical vesicles decreases as Δa
decreases.
-
Plasma Fusion Res. 11, 2401073 (2016).
-
-
An Intuitive Interface for Visualizing Numerical Data in a Head-Mounted Display with Gesture Control
-
Yuichi Tamura, Hiroaki Nakamura and Susumu Fujiwara
-
This study aims to create an interface for visualizing numerical data on
a head-mounted display (HMD) and introduce functions to allow control of
this visualization via hand gestures. HMDs have the advantage of providing
a user with a 360-degree field of view without taking up a lot of space.
However, it is difficult to control visualized numerical data intuitively
with this type of display because the user cannot see his/her own hand.
We therefore introduced functions allowing the user to control the virtual
scene with visualized virtual hands. We developed a system in which a
virtual menu is presented to the users and they can change the visualization
method by pushing virtual panels. The user can move freely in the visualized
virtual scene. Moreover, to help users share their thoughts, we have
introduced a drawing function that enables users to indicate points and
areas of interest in the visualized scene.
-
Plasma Fusion Res. 11, 2406060 (2016).
-
-
Melt memory of a spherulite nucleus formed through a seeding process in the crystal growth of isotactic polystyrene
-
Masato Hashimoto, Junko O'ishi, Sayoko Moriya and Susumu Fujiwara
-
The melt memory effect on the crystal growth of isotactic polystyrene is investigated using optical microscopy. After a spherulite is melted at a slightly higher temperature than the melting point and completely disappears, a polymer crystal nucleates and grows at the same position as the original spherulite when the temperature is decreased below the melting point. After the melting–recrystallization processes are repeated by raising and lowering the temperature, the melt memory effect becomes weak. This effect completely disappears at
∼40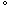 C
above the equilibrium melting point when the melt temperature is gradually increased during the melting–recrystallization processes. The formation of spherulite nuclei is promoted by the seeding process, which consists of the following consecutive procedures: quenching a sample to a temperature below the glass transition temperature and annealing the sample slightly above the glass transition temperature. At the initial stage, the number density of spherulites rapidly decreases with the cumulative melting time. After the initial stage, the number density of spherulites exponentially decays with the cumulative melting time and approaches a finite value. This asymptotic value decreases with the melt temperature.
C
above the equilibrium melting point when the melt temperature is gradually increased during the melting–recrystallization processes. The formation of spherulite nuclei is promoted by the seeding process, which consists of the following consecutive procedures: quenching a sample to a temperature below the glass transition temperature and annealing the sample slightly above the glass transition temperature. At the initial stage, the number density of spherulites rapidly decreases with the cumulative melting time. After the initial stage, the number density of spherulites exponentially decays with the cumulative melting time and approaches a finite value. This asymptotic value decreases with the melt temperature.
-
Polymer J. 47, 481-486 (2015).
-
-
Molecular Dynamics Simulation of Phase Behavior in a Bolaamphiphilic Solution
-
Susumu Fujiwara, Takumi Miyata, Masato Hashimoto, Yuichi Tamura, Hiroaki Nakamura and Ritoku Horiuchi
-
The phase behavior of bolaamphiphilic solutions is studied by coarse-grained
molecular dynamics simulations of semiflexible bolaamphiphilic molecules
with explicit solvent molecules. Our simulations show that six kinds of
self-assembled structures (spherical micelles, worm-like micelles,
bicontinuous structure, hexagonal structure, plate-like micelles, and
lamellar structure) are obtained. It is established that, at low
concentrations, a plate-like micelle changes to worm-like micelles, and
then to spherical micelles as the hydrophilic interaction increases.
Conversely, at intermediate concentrations, a lamellar structure changes
to a bicontinuous structure; it then changes to worm-like micelles or
a hexagonal structure as the hydrophilic interaction increases. It is
also observed that the global orientational order parameter for the end
bonds of bolaamphiphilic molecules can be used to clearly distinguish
between the randomly-oriented structures (the spherical micelles, the
worm-like micelles, and the bicontinuous structure), the lamellar
structure, the hexagonal structure, and the plate-like micelles.
-
Plasma Fusion Res. 10, 3401029 (2015).
-
-
Molecular Dynamics Simulation of Micellar Shape Transition in
Amphiphilic Solutions
-
Susumu Fujiwara, Masato Hashimoto, Yuichi Tamura, Hiroaki Nakamura
and Ritoku Horiuchi
-
The micellar shape transition in amphiphilic solutions is studied by
coarse-grained molecular dynamics simulations of rigid amphiphilic
molecules with explicit solvent molecules. Our simulations show that
the dominant micellar shape changes from disc to cylinder, and then
to sphere as the hydrophilic interaction increases. We find that,
as the hydrophilic interaction increases, the potential energy decreases
monotonically even during the micellar shape transition, whereas
the slope of the potential energy decreases in a stepwise manner
in relation to the micellar shape transition. We also ascertained
that there exists a wide coexistence region in the intensity of
the hydrophilic interaction between a cylinder and a sphere, whereas
the coexistence region between a cylinder and a disc is very narrow.
-
Plasma Fusion Res. 9, 3401067 (2014).
-
-
Nucleation and polymorphism of trans-1,4-polyisoprene containing copper
phthalocyanine
-
Tomoya Tsuboi, Masashi Harada, Kei Ishii, Susumu Fujiwara and Takashi Itoh
-
The effect of α- or β-copper
phthalocyanine (CuPc) as a nucleating agent during the crystallization of
trans-1,4-polyisoprene (TPI) was evaluated using polarized optical
microscopy (POM), differential scanning calorimetry (DSC) and wide angle
X-ray diffraction (WAXD). Pure TPI crystallizes into either a high-melting
crystal form (HMF) or a low-melting crystal form (LMF), depending on the
cooling condition of the melt. The HMF melts at
∼60C and
LMF typically melts at
∼50C.
Observation by POM showed that the LMF is generated at the interface between
TPI and either α- or β-CuPc,
selectively, when crystallized at
38C.
In DSC measurements, the TPI composites containing
α- or β-CuPc showed a higher
crystallization temperature than that of pure TPI during the melt cooling
process. Such results provided evidence that the
α- or β-CuPc acted as a nucleating
agent for TPI. The data obtained from WAXD proved that the HMF was generated
mainly when pure TPI was cooled from the melt at a rate of
5C min−1,
while the LMF was generated predominantly when the
TPI/α- or
β-CuPc composites were crystallized under the same cooling conditions
as those used for pure TPI. These results led to the conclusion that
α- and β-CuPc acted as phase
selective nucleating agents for LMF-TPI.
-
Polymer J. 45(9), 915-920 (2013).
-
-
One-, Two-, and Three-Dimensional Hopping Dynamics
-
Keiko M. Aoki, Susumu Fujiwara, Kiyoshi Sogo, Shuhei Ohnishi and
Takenori Yamamoto
-
Hopping dynamics in glass has been known for quite a long time. In contrast,
hopping dynamics in smectic-A (SmA) and hexatic smectic-B (HexB) liquid
crystals (LC) has been observed only recently.
The hopping in SmA phase occurs among the smectic layers (one-dimensionally),
while hopping in HexB phase occurs inside the layers (two-dimensionally).
The hopping dynamics in SmA and HexB liquid crystal phases is investigated
by parallel soft-core spherocylinders, while three-dimensional hopping
dynamics in inherent glassy states is investigated by systems of
Weeks-Chandler-Andersen (WCA) spheres.
The temperature dependence of diffusion coefficients of hopping in SmA phase
can be described by the Arrhenius equation characteristic of activation process.
In HexB LC phase, the diffusion coefficients saturate at higher temperatures.
In a system of WCA spheres, the values and temperature dependence of diffusion
coefficients depend on the observed states.
-
Crystals 3(2), 315-332 (2013).
-
-
Micellar Shape Change in Amphiphilic Solution: A Molecular Dynamics Study
-
Susumu Fujiwara, Masato Hashimoto, Takashi Itoh and Ritoku Horiuchi
-
Micellar shape change in amphiphilic solution after sudden change in the
solvophilic interaction is studied by molecular dynamics simulations of
coarse-grained, rigid amphiphiles.
Our simulations demonstrate that, after sudden increase in intensity of
the solvophilicity, the disc micelle transforms into cylindrical or
spherical micelles immediately.
In contrast, spherical micelles coalesce into a disc micelle in a
stepwise manner after sudden decrease in intensity of the solvophilicity.
-
Chem. Lett. 41(10), 1038-1040 (2012).
-
-
Dissipative Particle Dynamics Simulation of Phase Behavior in
Bolaamphiphilic Solution
-
Ryōen Shirasaki, Yuta Yoshikai, Hu-Jun Qian, Susumu Fujiwara, Yuichi Tamura
and Hiroaki Nakamura
-
We study the phase behavior of bolaamphiphilic solution performing the
dissipative particle dynamics simulations of coarse-grained bolaamphiphilic
molecules with explicit solvent molecules.
Our simulations show that there are six kinds of phases: isotropic micellar,
micellar, rod-shaped micellar, hexagonal, network-structure and lamellar.
The network-structure and the lamellar phases disappear when the restoring
potential against the bending of bolaamphiphilic molecules in our simulation
model is excluded; and the isotropic micellar and the hexagonal phases
disappear when the restoring potential is included.
This suggests that the bending potential is important in the formation
of the higher-ordered structures by the bolaamphiphilic molecules.
-
Plasma Fusion Res. 6, 2401116 (2011).
-
-
Design Support System with Haptic Feedback and Real-Time Interference Function
-
Yuichi Tamura, Koji Ukita, Naoki Mizuguchi and Susumu Fujiwara
-
When we design and construct a large-scale device, it is very important to
confirm the interference among its parts.
We might need to confirm not only the interference among the parts that
are designed at the start but also the interference with some parts that
are added after construction.
However, sometimes even on using 3D CAD, we cannot detect the interference
or the collision among parts, particularly when these parts form a complex
3D shape.
On the other hand, virtual reality devices have been used in various
fields such as design support systems; however, real-time collision
detection among complex parts has been difficult to achieve.
We constructed a system that can detect collision and interference
in real time in a virtual reality system.
This engine can detect interference between polygons.
This enables to calculate more accurately than voxel-based detection engine.
Moreover, we propose a dynamic interference vector for removing interference.
This vector is defined as a local minimum vector which removes the
interference from the same edge where the contact starts.
This method enables to prevent an object moves discontinuously,
when the interference is removed. Finally, we introduced an example of
using this system for assembling parts.
-
Plasma Fusion Res. 6, 2406061 (2011).
-
-
Molecular Dynamics Simulation of Micellar Shape Change in Amphiphilic Solution
-
Susumu Fujiwara, Takashi Itoh, Masato Hashimoto, Yuichi Tamura,
Hiroaki Nakamura and Ritoku Horiuchi
-
Micellar shape change in an amphiphilic solution is investigated by
a molecular dynamics simulation of coarse-grained semiflexible amphiphilic
molecules with explicit solvent molecules.
Our simulations show that a cylindrical micelle is obtained at small
molecular rigidity while a disc-shaped micelle appears at large molecular
rigidity.
We find that most chains are in an extended conformation at large
molecular rigidity whereas the fraction of the chains in a bent
conformation becomes large at small molecular rigidity.
It is also ascertained that the micellar shape starts to change
immediately after sudden increase of the molecular rigidity
while an induction time is needed to change the micellar shape
after sudden decrease of the molecular rigidity.
This result can be qualitatively explained by considering the
bond-bending potential energy and the conformational entropy of the
amphiphilic molecules.
-
Plasma Fusion Res. 6, 2401040 (2011).
-
-
Bracelet-shaped thermal display for representing numerical data
-
Yuichi Tamura, Susumu Fujiwara, Tomohiro Umetani and Hiroaki Nakamura
-
A thermal display, a type of haptic display, is effective for providing
intuitive information about temperature. In many thermal display studies,
users have assumed sitting positions when using these devices.
However, their use in a large-scale virtual-reality system requires
users to be in a standing position, as they generally observe
three-dimensional (3D) objects while standing or walking around.
Thus, we developed thermal displays that are suitable for large-scale
virtual-reality systems.
From another standpoint, in scientific visualization, response time
is very important for observing physical phenomena, especially for
dynamic numerical simulation.
One way to optimize this parameter is to provide two types of thermal
information: the rate of thermal change, and the actual temperature.
To this end, we propose a bracelet-shaped thermal display with
three Peltier elements that can provide both types of information.
Finally, we present an example of visualizing and haptizing
the result of a molecular dynamics simulation.
-
J. Electron. Mater. 40(5), 823-829 (2011).
-
-
Molecular dynamics simulation for phase behavior of amphiphilic solution
-
Susumu Fujiwara, Daiki Funaoka, Takashi Itoh and Masato Hashimoto
-
The phase behavior of amphiphilic solution is investigated by molecular
dynamics simulation of amphiphilic rigid dimers with explicit solvent
molecules. Our simulations show that three kinds of phases (isotropic
micellar, hexagonal and lamellar phases) are formed at a lower temperature
by quenching from a random configuration of amphiphilic molecules in
solution at a higher temperature. It is ascertained that an isotropic
micellar phase changes into a hexagonal phase, and then into a lamellar
phase as the amphiphilic concentration increases. It is also found that
the global orientational order parameter can be used to distinguish
these three kinds of phases. From the detailed analyses of the phase
behavior, it is concluded that the hydrophilic repulsion plays an
important role in the formation of the hexagonal phase while the
hydrophobic attraction plays a crucial role in the formation of
the lamellar phase.
-
Computer Physics Communications 182(1), 192-194 (2011).
-
-
Comparison of Hydrogen Adsorption on Diamond and Graphite Surfaces
-
Hiroaki Nakamura, Atsushi Ito, Seiki Saito, Yuichi Tamura, Susumu Fujiwara,
Noriyasu Ohno and Shin Kajita
-
By a classical molecular dynamics (CMD) simulation with a modified Brenner's
reactive empirical bond-order (REBO) potential, we found that
graphite with zigzag (1010)
and armchair (1120)
edge states is destroyed
more easily than the other structures, i.e., graphite with (0001) surface,
and diamond with the (100), (111), (120), and (110) surfaces.
Experimental results indicated that graphite is eroded under hydrogen
atom injection with Ein = 0.3 eV, and that diamond is not eroded
under the same conditions.
Our simulation results are consistent with the experimental results.
We also reveal the temperature and saturation dependence of
the surface structure of the carbon crystals.
-
Plasma Fusion Res. 5, S2072 (2010).
-
-
Haptization of molecular dynamics simulation with thermal display
-
Yuichi Tamura, Susumu Fujiwara and Hiroaki Nakamura
-
Thermal display, which is a type of haptic display, is effective in providing intuitive information of temperature.
However, in many studies, the user has assumed a sitting position during the use of these devices.
In contrast, the user generally watches 3D objects while standing and walking around in large-scale virtual reality system, In addition, in scientific visualization, the response time is very important for observing physical phenomena, especially for dynamic numerical simulation.
One solution is to provide two types of thermal information: information about the rate of thermal change and information about the actual temperature.
We propose a thermal display with two Peltier elements which can show above two pairs of information and the result (for example energy and temperature, as thermal information) of numerical simulation.
Finally, we represent an example of visualizing and haptizing the result of molecular dynamics simulation.
-
Plasma Fusion Res. 5, S2107 (2010).
-
-
Effect of Molecular Rigidity on Micelle Formation in Amphiphilic Solution
-
Susumu Fujiwara, Takashi Itoh, Masato Hashimoto, Hiroaki Nakamura and
Yuichi Tamura
-
Micelle formation in an amphiphilic solution is investigated by
a molecular dynamics simulation of coarse-grained semiflexible
amphiphilic molecules with explicit solvent molecules.
Our simulations show that the micellar shape changes from a cylinder
to a disc as the intensity of the molecular rigidity increases.
We find that the radius of gyration of the cylindrical micelle is
larger than that of the disc-shaped micelle for small molecular rigidity,
although the radius of gyration is almost steady
even during the transition between a cylinder and a disc
for large molecular rigidity.
This indicates that a cylindrical micelle formed at
small molecular rigidity is more anisotropic than the one obtained
at large molecular rigidity.
We also ascertained that a cylindrical micelle and a disc-shaped micelle
coexist dynamically over a certain molecular rigidity range.
-
Plasma Fusion Res. 5, S2114 (2010).
-
-
Correlated Anomalous Diffusion - Random walk and Langevin equation -
-
Kiyoshi Sogo, Yoshiaki Kishikawa, Shuhei Ohnishi, Takenori Yamamoto,
Susumu Fujiwara and Keiko M. Aoki
-
A random walk model is formulated and examined which gives the correlated
anomalous diffusion found in molecular dynamics simulations. The mean square
displacement (MSD) shows a logarithmic behavior in one dimension.
Corresponding Langevin equation is constructed by solving the inverse
problem which gives a procedure to derive random impulse correlation from
MSD function.
-
J. Math. Phys. 51(3), 033302 (2010).
-
-
Molecular dynamics simulations for structure formation of polymers
and self-assembly of amphiphilic molecules
-
Susumu Fujiwara, Masato Hashimoto and Takashi Itoh
-
Molecular dynamics simulations were carried out to study the structure
formation of polymers and the self-assembly of amphiphilic molecules.
In particular, the structure formation of an isolated single polymer
chain, isolated short chain molecules, and a single polymer chain in
solution, and the spontaneous micelle formation in amphiphilic solution
were investigated. From the detailed analyses of the structure formation
processes, two characteristic features, the stepwise energy relaxation
and the dynamic coexistence, are clarified as those common to the
nonequilibrium dynamics in chain-like molecules and amphiphiles
systems. We believe that these two findings will provide a key to the
research on the universality of the nonequilibrium dynamics.
-
Kobunshi Ronbunshu 66(10), 396-405 (2009) [in Japanese].
-
-
Novel Primary Dispersion in Viscoelastic Behavior of Ferroelectric Nylon 6
-
Tsutomu Takahashi, Takashi Itoh, Susumu Fujiwara and Masato Hashimoto
-
As-quenched ferroelectric amorphous nylon 6 shows a new intense primary
relaxation (α') peak for
tan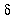 and E" around 300 K.
Activation energy of α' relaxation was
estimated to be about 150 kcal/mol, ca. three times that of
α relaxation. It suggests that loose
molecular packing in the ferroelectric amorphous phase makes the
large-scale molecular motion possible, when rotational motion of
amide groups is activated and D-E hysteresis loops
can be observed.
and E" around 300 K.
Activation energy of α' relaxation was
estimated to be about 150 kcal/mol, ca. three times that of
α relaxation. It suggests that loose
molecular packing in the ferroelectric amorphous phase makes the
large-scale molecular motion possible, when rotational motion of
amide groups is activated and D-E hysteresis loops
can be observed.
-
Polymer J. 41(5), 354 - 355 (2009).
-
-
Molecular dynamics simulation of amphiphilic molecules in solution:
Micelle formation and dynamic coexistence
-
Susumu Fujiwara, Takashi Itoh, Masato Hashimoto and Ritoku Horiuchi
-
The micelle formation and the dynamic coexistence in amphiphilic
solution are investigated by molecular dynamics simulation of
coarse-grained rigid amphiphilic molecules with explicit solvent
molecules. Our simulations show that three kinds of isolated micelles
(disc, cylindrical, and spherical micelles) are observed at a lower
temperature by quenching from a random configuration of amphiphilic
molecules in solution at a higher temperature. The micellar shape
changes from a disc into a cylinder, and then into a sphere as
the hydrophilic interaction increases whereas it is not so sensitive
to the variation of the hydrophobic interaction. This fact indicates
that the hydrophilic interaction plays an important roll in
determining the micellar shape in the range of the interaction
parameters used. It is also found that, in a certain interaction
parameter range, two kinds of micellar shapes coexist dynamically.
From the detailed analyses of the dynamic coexistence, it is
ascertained that the dynamic coexistence of a cylindrical micelle
and a spherical micelle accompanies the coalescence and fragmentation
of micelles while that of a disc micelle and a cylindrical micelle
does not, but exhibits the continuous change between them.
-
J. Chem. Phys. 130(14), 144901 (2009).
-
-
Freezing of High-Temperature Phase in Vinylidene Fluoride/Trifluoroethylene
Copolymer Crystals on Vacuum-Evaporated Metal Surfaces
-
Thuy-Trang Hua, Takashi Itoh, Jian-An HOU, Susumu Fujiwara and Masato Hashimoto
-
Vinylidene fluoride/trifluoroethylene copolymer thin films cast from dilute
cyclohexanone solution on vacuum-evaporated Pt, Al, Cu, and Au were annealed
at 420K for 1 h.
Transmission electron microscopic observation revealed that the hightemperature
phase (HP) of the copolymers was frozen even at room temperature with molecular
chains normal to the substrate after the annealing.
The maximum lattice mismatch was 3.3% between the (001) plane of the HP of
the copolymers and the {111} or {110} plane of the Pt crystals.
Coulomb’s interaction between the copolymer molecules and metallic atoms is
suggested to have an important role in causing such phenomena.
-
Jpn. J. Appl. Phys. 46(47), L1170 - L1172 (2007).
-
-
Superlattice Epitaxy of Vinylidene Fluoride/Trifluoroethylene Copolymer
Crystals on Highly Oriented Pyrolytic Graphite
-
Thuy-Trang Hua, Takashi Itoh, Susumu Fujiwara and Masato Hashimoto
-
Vinylidene fluoride/trifluoroethylene (VDF/TrFE) copolymer crystals were grown
from the dilute solution on cleaved (0001) surface of highly oriented pyrolytic
graphite (HOPG). Using transmission electron microscopy, the orientational
relation between the copolymer crystals and cleaved (0001) surface of HOPG is
clarified, as well as the supperlattice matching.
Electron diffraction (ED) patterns with 6-fold symmetry were observed for
the copolymer crystals with VDF molar contents of 59, 65, and 71%.
It suggests that a rectangular array of
(201)
plane for the high-temperature phase (HP) of the copolymer crystal faces
HOPG(0001) plane, from which the molecular chains tilt by 46.7°.
The ED pattern with such 6-fold symmetry is attributed to superposition of
diffractions from the HP crystallites with three different orientations on HOPG.
Such results suggest that epitaxial growth of HP occurred and it was frozen or
stabilized even at room temperature due to epitaxial effect or interaction
between copolymer molecules and substrate atoms.
-
J. Phys. Soc. Jpn. 76(12), 124604 (2007).
-
-
Molecular dynamics simulation of micelle formation in amphiphilic solution
-
Susumu Fujiwara, Takashi Itoh, Masato Hashimoto and Yuichi Tamura
-
The micelle formation in amphiphilic solution is investigated by
molecular dynamics simulation of coarse-grained rigid amphiphiles
with explicit solvent molecules.
In our simulation model, the intensity of the hydrophilic interaction
and the hydrophobic interaction can be varied independently.
Our simulations show that various kinds of micellar structures are
formed at a lower temperature by quenching from a random configuration
of amphiphilic molecules in solution at a higher temperature.
The micellar shape changes from a disc (bilayer) into a cylinder, and then
into a sphere as the intensity of the hydrophilic interaction increases.
It is also found that the micelle formation proceeds in a stepwise
fashion through the coalescence of smaller micelles.
From the analysis of the orientational order for the amphiphilic molecules,
it is concluded that the orientational order parameters can be used
to distinguish the micellar shapes clearly.
-
Mol. Simul. 33(1-2), 115-119 (2007).
-
-
Molecular dynamics simulation of self-organization in amphiphilic solution
-
Susumu Fujiwara, Masato Hashimoto and Takashi Itoh
-
The micelle formation in amphiphilic solution is investigated by
means of a molecular dynamics simulation of coarse-grained
amphiphilic molecules with explicit solvent molecules.
A random configuration of amphiphilic molecules in solution at high
temperature is quenched to a lower temperature.
Our simulations show that the micellar shapes change
from a cylindrical micelle to a planar bilayer
as the number density increases.
At higher densities, we also find the following characteristic features:
(1) The potential energy relaxes in a stepwise manner.
(2) The radius of gyration Rg of the largest
micelles increases with time in a stepwise fashion.
(3) The sharp bumps in Rg occur during coalescence
of micelles.
-
J. Plasma Phys. 72(6), 1011-1014 (2006).
-
-
Molecular Dynamics Simulation for Structure Formation
of Single Polymer Chain in Solution
-
Susumu Fujiwara, Masato Hashimoto, Takashi Itoh and Hiroaki Nakamura
-
The structure formation of a single polymer chain in solution with
explicit solvent molecules is investigated by molecular
dynamics simulation.
The orientationally ordered structure is formed at a low
temperature by quenching from a random conformation at
a high temperature.
The growth of the global and local orientational order proceeds
in a stepwise manner at T 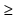 350 K, whereas it proceeds
in a gradual manner at T = 300 K.
From the detailed analyses of the parallel ordering process,
it is found that a conformational change of the polymer chain
occurs at first, and then parallel ordering starts to take place.
In comparison with the simulation results of an isolated polymer
chain in vacuum, it is ascertained that the stem length of the
orientationally ordered structure formed in solution becomes 2-3
times longer than that formed in vacuum.
350 K, whereas it proceeds
in a gradual manner at T = 300 K.
From the detailed analyses of the parallel ordering process,
it is found that a conformational change of the polymer chain
occurs at first, and then parallel ordering starts to take place.
In comparison with the simulation results of an isolated polymer
chain in vacuum, it is ascertained that the stem length of the
orientationally ordered structure formed in solution becomes 2-3
times longer than that formed in vacuum.
-
J. Phys. Soc. Jpn. 75(2), 024605 (2006).
-
-
Thermal, Structural and Ferroelectric Properties of Amorphous Phases
in Quenched Nylon 6 Film
-
Teruaki Yanagisawa, Takashi Itoh, Yasuo Saruyama and Susumu Fujiwara
-
Three types of amorphous phases (ferroelectric metastable phase A with
large remanent polarization, ferroelectric metastable phase B with small
remanent polarization and paraelectric stable phase C) are identified in
as-quenched and/or annealed nylon 6 films on the basis of the thermal
data obtained by light-modulated differential scanning calorimetry
(LMDSC) as well as X-ray data, which show different glass transition
temperatures and d-spacings. The irreversible exothermic anomaly
at 328 K observed through heating process in the conventional DSC is
attributed to transition from phases A and/or B to nematic phases.
The transition from phase A to phase B is considered to complete after
15 min anneling at 320 K on the basis of the LMDSC data, which corresponds
to abrupt diminution of the remanent polarization by the correspondent
anneling condition. Such results suggest that ferroelectricity of nylon 6
film is mainly attributed to phase A (frozen state of the melt) formed in
quenched nylon 6 thin film.
-
J. Phys. Soc. Jpn. 73(10), 2763-2767 (2004).
-
-
Phase Transition in Even-Even Nylon Crystals
-
Hiroya Ishikawa, Takashi Itoh, Masato Hashimoto and Susumu Fujiwara
-
Temperature dependence of crystal structures is examined with X-ray
diffraction at normal pressure or elevated pressures for
solution-grown nylons with octamethylene moiety between amino
or carboxy groups (nylons 82, 410, 610, 810). Although octamethylene
moiety between amino groups has the potential to cause the phase
transition, the low melting point veils the transition appearance.
Considering this comprehensively with previous results, it is concluded
for all even-even nylons, that the phase transition in the two-dimensional
methylene layer system can be experimentally observed only in nylon 6Y
(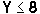 ) crystals at normal and/or high pressures, where the hexamethylene
moiety between amino groups plays an intrinsic role. The so-called
`Brill transition' widely reported for melt-crystallized nylons
is not a phase transition but an abrupt thermal expansion.
) crystals at normal and/or high pressures, where the hexamethylene
moiety between amino groups plays an intrinsic role. The so-called
`Brill transition' widely reported for melt-crystallized nylons
is not a phase transition but an abrupt thermal expansion.
-
J. Phys. Soc. Jpn. 73(2), 303-306 (2004).
-
-
Gradient Pattern Analysis of Structural Dynamics: Application to
Molecular System Relaxation
-
Reinaldo R. Rosa, Marcia R. Campos, Fernando M. Ramos,
Nandamudi L. Vijaykumar, Susumu Fujiwara and Tetsuya Sato
-
This paper describes an innovative technique, the gradient pattern
analysis (GPA), for analysing spatially extended dynamics. The
measures obtained from GPA are based on the spatio-temporal correlations
between large and small amplitude fluctuations of the structure
represented as a dynamical gradient pattern. By means of four gradient
moments it is possible quantify the relative fluctuations and scaling
coherence at a dynamical numerical lattice and this is a set of proper
measures of the pattern complexity and equilibrium. The GPA technique
is applied for the first time in 3D-simulated molecular chains with the
objective of characterizing small symmetry breaking, amplitude and phase
disorder due to spatio-temporal fluctuations driven by the spatially
extended dynamics of a relaxation regime.
-
Brazilian J. Phys. 33(3), 605-610 (2003).
-
-
Structure Formation of a Single Polymer Chain in Solution: A Molecular
Dynamics Study
-
Susumu Fujiwara and Tetsuya Sato
-
Molecular dynamics simulations are carried out to study the structure
formation of a single polymer chain in solution. A random conformation
of a polymer chain in solution at high temperature (550 K) is quenched
to several lower temperatures (300, 350, and 400 K). Our simulations
show the following characteristics: 1) At lower quenching temperature
(300 K), local orientationally ordered domains are formed first, and
they grow to be a folded orientationally ordered structure. 2) At a
higher quenching temperature (400 K), a toroidal structure can be formed.
3) At an intermediate quenching temperature (350 K), a toroidal structure
can be formed before a folded orientationally ordered structure is formed.
The last characteristic indicates that an intermediate toroidal structure
at 350 K is in a metastable state. We also find that it depends on the
initial conformation of a polymer chain, whether a toroidal structure is
formed or not.
-
J. Macromol.Sci. - Physics B42(3&4), 455-466 (2003).
-
-
Molecular dynamics simulation of a single polymer chain in vacuum and
in solution
-
Susumu Fujiwara and Tetsuya Sato
-
Molecular dynamics simulations are carried out to study the structure
formation of a single polymer chain in vacuum and in solution. We find
that, in both cases, the folded orientationally ordered structure is
formed at a low temperature by quenching from a random configuration
at a higher temperature. It is also found that the stem length of the
folded orientationally ordered structure formed in solution is longer
than that formed in vacuum. This result is caused by the fact that the
persistence length of a polymer chain in solution becomes longer than
that in vacuum.
-
Computer Physics Communications 147, 342-345 (2002).
-
-
Dynamics of orientationally ordered domains in a short chain-molecule system:
Size dependence of domain oscillation
-
Hiroaki Nakamura, Susumu Fujiwara and Tetsuya Sato
-
It was reported, in our previous works, that two orientationally ordered
domains of short chain molecules move collectively as if they were rigid
bodies in spite of the non-bonded short-range interaction potential
(Lennard-Jones potential) among chain molecules. In this paper, in order
to investigate the rigidity of the domain of short chain molecules in
further detail, molecular dynamics simulations are performed for the
following four case combinations of domains: the case I, II, III and IV
are (61+29), (61+61), (124+61) and (124+124) chain molecule combinations,
respectively. From these simulations, the size dependence of the domains'
motion is demonstrated. Estimating an oscillation period
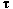 for each case,
it is found that
for each case,
it is found that 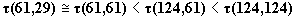 .
This relation is a peculiar property of orientationally ordered domains in
a short chain-molecule system.
.
This relation is a peculiar property of orientationally ordered domains in
a short chain-molecule system.
-
Computer Physics Communications 147, 346-349 (2002).
-
-
Molecular dynamics study of structure formation of a single polymer
chain by cooling
-
Susumu Fujiwara and Tetsuya Sato
-
Structure formation of a single polymer chain with 500 methylene groups
is investigated by means of a molecular dynamics simulation.
We find that the folded orientationally ordered structure is formed
at a low temperature by gradual stepwise cooling or by quenching
from a random configuration at a higher temperature.
It is also found that the global orientational order grows
in a gradual manner in the case of gradual stepwise
cooling, whereas it grows in a stepwise manner in the case
of quenching.
-
Computer Physics Communications 142, 123-126 (2001).
-
-
Dynamical Process of Coalescence of Domains in a Short Chain-molecule System
-
Hiroaki Nakamura, Susumu Fujiwara and Tetsuya Sato
-
As the fundamental process of structure formation for short chain molecules,
the coalescence of two orientationally ordered domains are investigated by
numerical simulation. Each domain consists of 61 chain molecules, each of which
consists of 20 CH2 groups. One domain is tilted against another one
with a certain angle. Then, the potential energy excited by tilting makes each
domain rotate gradually. In this process, it is demonstrated that domains move
collectively as if they were rigid bodies in spite of the non-bonded short-range
interaction potential (Lennard-Jones potential) among chain molecules. From
analysis of oscillation period of each potential well, it is concluded that
the rigidity of domain derives its origin from the fact that the Lennard-Jones
potential interaction of the unit hexagonal cell is strengthened to the order
of the bonded interaction by packing.
-
Computer Physics Communications 142, 127-130 (2001).
-
-
Virtual reality system to visualize and auralize numerical simulation data
-
Yuichi Tamura, Akira Kageyama, Tetsuya Sato, Susumu Fujiwara and
Hiroaki Nakamura
-
One of the most practical objectives to use the virtual reality (VR) system in
science is to make it easy to intuitively percept complex physical phenomena.
We developed a VR system, called CompleXcope, which can represent "real" 3D
visual and aural environment. Since attractive physical phenomena are often so
complex and tangled, the VR system with 3D-sound functions is really useful for
the quick comprehension of phenomena. We also show an example of visualizing
and auralizing the molecular dynamic simulation results.
-
Computer Physics Communications 142, 227-230 (2001).
-
-
Rigidity of Orientationally Ordered Domains of Short Chain Molecules
-
Hiroaki Nakamura, Susumu Fujiwara and Tetsuya Sato
-
By molecular dynamics simulation, discovered is a strange rigid-like
nature for a hexagonally packed domain of short chain molecules.
In spite of the non-bonded short-range interaction potential
(Lennard-Jones potential) among chain molecules, the packed domain gives
rise to an apparent global moment of inertia. Accordingly, as two domains
encounter obliquely, they rotate so as to be parallel to each other keeping
their overall structures as if they were rigid bodies.
-
J. Phys. Soc. Jpn. 70(4), 943-946 (2001).
-
-
Structure formation of a single polymer chain. I: Growth of trans
domains
-
Susumu Fujiwara and Tetsuya Sato
-
Molecular dynamics simulations are carried out to study structure
formation of a single polymer chain with 500 CH2 groups.
Our simulations show that the orientationally ordered structure
is formed at a low temperature both by gradual stepwise cooling and
by quenching from a random configuration at a higher temperature.
The growth of the global orientational order proceeds
in a gradual manner in the case of gradual stepwise cooling,
whereas it proceeds in a stepwise manner in the case of quenching.
The latter feature endorses the previously proposed hypothetical grand
view of self-organization [e.g. T. Sato, Phys. Plasmas 3, 2135
(1996)]: when a system is driven far from equilibrium, it will evolve
to a more stable state in a stepwise fashion irrespective of its
fundamental interaction forces.
From the microscopic analysis of the structure formation process,
we find the following characteristic features:
(i) In the case of gradual stepwise cooling, the global orientational
order grows gradually through the incorporation of small trans
domains and the surrounding trans segments into the largest
trans domain.
(ii) In the case of quenching, the growth of the orientational order
is either due to the incorporation of small trans domains and
the surrounding trans segments into the largest trans
domain or due to the elongation of the trans segments in the
largest trans domain.
-
J. Chem. Phys. 114(14), 6455-6463 (2001).
-
-
Aural-Visual Virtual Representation System for Numerical Simulation Data
-
Yuichi Tamura, Tetsuya Sato, Akira Kageyama, Susumu Fujiwara and
Hiroaki Nakamura
-
One of the most practical objectives to use the virtual reality system
in science is to make it easy to intuitively percept complex physical
phenomena. We developed a virtual reality system, called CompleXcope,
which can represent "real" 3D visual environment. Since attractive
physical phenomena are often so complex and tangled, the virtual
reality system with 3D-sound functions is really useful for the quick
comprehension of phenomena. We have recently added a sound function to
the CompleXcope. In this paper we present examples produced by this
aural-visual virtual reality system "CompleXcope".
-
Transactions of the Virtual Reality Society of Japan
5(3), 943-948 (2000) [in Japanese].
-
-
Molecular Dynamics Simulation of Chain-molecule Systems
--- Distribution of Conformational Defects ---
-
Susumu Fujiwara and Tetsuya Sato
-
Molecular dynamics simulations are carried out to study the
conformational defects in the orientationally ordered structures
of short chain molecules and a single polymer chain.
Our simulations show that the orientationally ordered structure
is formed from a random configuration by cooling.
There exist no fold surfaces in the orientationally ordered
structure of short chain molecules while fold surfaces exist in
the ordered structure of a single polymer chain.
From detailed analyses of the conformational defects, we find
that, in the case of short chain molecules, the double gauche
defects with the same sign (...tg+g+t...)
are predominantly located at the chain ends and the kink defects
(...tg+tg-t...), which do not cause serious
deformation of chain molecules, can exist even at the chain
interiors.
On the other hand, in the case of a single polymer chain,
several types of the conformational defects which give rise to
large deformation of a polymer chain, such as the double
gauche defects with the opposite sign
(...tg+g-t...), can be observed in the
fold surfaces.
-
Prog. Theor. Phys. Supplement No.138, 342-347 (2000).
-
-
Structure Formation in a Short Chain-molecule System:
A Molecular Dynamics Study
-
Susumu Fujiwara and Tetsuya Sato
-
The structure formation of 100 short chain molecules, each of which
consists of 20 CH2 groups, is investigated by means of a
molecular dynamics simulation.
The orientationally ordered structure is formed at a lower temperature
by a sudden cooling from a random configuration at a higher temperature.
It is also found that the growth of the local ordered clusters proceeds
in a stepwise fashion.
-
J. Plasma Fusion Res. SERIES Vol.2, 498-500 (1999).
-
-
Molecular dynamics simulation of structure formation of
short chain molecules
-
Susumu Fujiwara and Tetsuya Sato
-
Molecular dynamics simulations are carried out to study
the structure formation of 100 short chain molecules,
each of which consists of 20 CH2 groups.
Our simulations show that the orientationally ordered structure
is formed from a random configuration by quenching.
The global orientational order starts to increase suddenly after
a certain duration and grow in a stepwise fashion afterwards.
This behavior is also found in the growth process of the local
orientationally-ordered domains.
It is found from the microscopic analysis of the structure
formation process that parallel ordering of chain molecules
starts to occur after the chain molecules stretch
to some extent.
From the analysis of the obtained orientationally ordered
structure and the molecular mobility, we also find the
following characteristic features:
(i) The chain molecules are packed hexagonally at 400 K
and the transition from the hexagonal phase toward the
orthorhombic phase takes place as the temperature decreases.
(ii) The gauche bonds in the same chain molecule tend to
form gauche pairs.
The gauche pairs with the same sign form the double
gauche defects and those with the opposite sign
form the kink defects.
(iii) In the hexagonal phase, the chain molecules become
longitudinally mobile.
This result, which is obtained by the microscopic analysis of
the chain motion, is the microscopic evidence to confirm
the existence of the chain sliding diffusion in the hexagonal
phase which underlies the sliding diffusion theory of polymer
crystallization proposed by Hikosaka
[Polymer 28, 1257 (1987); 31, 458 (1990)].
-
J. Chem. Phys. 110(19), 9757-9764 (1999).
-
-
MOLECULAR DYNAMICS STUDY OF THE STRUCTURAL FORMATION
OF SHORT CHAIN MOLECULES: STRUCTURE AND MOLECULAR MOBILITY
-
Susumu Fujiwara and Tetsuya Sato
-
By carrying out the molecular dynamics simulations of 100 short
chain molecules, each of which consists of 20 CH2 groups,
we show that the orientationally ordered structure is formed
at low temperature by a sudden cooling from a random configuration
at high temperature.
The essentially extended chains form a monolayer structure.
The ratio of the lattice constants a/b takes the hexagonal value
31/2 at 400 K and decreases as the temperature decreases.
From detailed analysis of the local orientational order, it is found
that the growth of the local ordered clusters proceeds
in a stepwise fashion.
From the analysis of the molecular mobility, we find that
the longitudinal chain motion increases dramatically with increasing
temperature while the transverse chain motion is not so sensitive
to the temperature variation.
-
Mol. Simul. 21, 271-281 (1999).
-
-
Molecular Dynamics Simulation of Structural Formation of
Short Polymer Chains
-
Susumu Fujiwara and Tetsuya Sato
-
Molecular dynamics simulations are carried out to study
the structural formation of 100 short polymer chains,
each of which consists of 20 CH2 groups.
Our simulations show that the orientationally ordered structure
at low temperature is formed from a random structure
at high temperature by a sudden cooling.
The essentially extended chains form a monolayer structure
with a hexagonal packing.
From detailed analyses of the local and global orientational order,
it is found that the formation of the global orientational order
as well as the growth of the local ordered regions proceeds stepwise.
-
Phys. Rev. Lett. 80(5), 991-995 (1998).
-
-
Molecular dynamics simulations of structural formation of a single
polymer chain: Bond-orientational order and conformational defects
-
Susumu Fujiwara and Tetsuya Sato
-
The structural formation of a single polymer chain with 500 CH2
groups is studied by the molecular dynamics simulations. Our simulations show
that the bond-orientationally ordered structure at low temperatures is
formed from a random-coil structure at high temperatures by a gradual
stepwise cooling. From the radii of gyration and the bond-orientational
order parameters, it is found that the anisotropy of a polymer chain
also grows during the growth of the bond-orientational order. In the
bond-orientationally ordered structure at low temperatures, 16 stems form
a structure with deformed hexagonal symmetry and the stems in the outer
layer have a tilted configuration. Furthermore, the gauche states
are localized in the fold surface and the conformational states in the fold
surface change more readily than those in the orientationally ordered
reagion.
-
J. Chem. Phys. 107(2), 613-622 (1997).
-
-
Model of anomalous relaxation in supercooled liquids:
random walk in fractal space and time
-
Fumiko Yonezawa, Susumu Fujiwara and Sohei Gomi
-
Anomalous structural relaxation observed in the supercooled liquids has
been one of the most exciting problems for the last decade. Toward the full
understanding of this phenomenon from the microscopic viewpoints, we
report in this article the results of our studies on the several random
walk models, i.e., random walk on fractal and non-fractal structures and the
fractal time random walk model. It is found that the relaxation becomes
of the Cole-Cole type, a famous empirical law, when the models have the
fractal nature while the stretched-exponential type relaxation is observed
in non-fractal structures. This work indicates that the concept of
`fractal' introduced with mathematical mind has important relevance to
realistic physical systems.
-
J. Non-Cryst. Solids 205-207, 884-887 (1996).
-
-
Anomalous relaxation in fractal and disordered systems
-
Susumu Fujiwara and Fumiko Yonezawa
-
Anomalous relaxation was studied by the Monte Carlo, MC, simulations of a
random walk in fractal and disordered structures. The calculations of the
relaxation functions and the complex susceptibilities show that the
anomalous relaxation is of the Cole-Cole type in fractal structures while it
is of the stretched-exponential type in non-fractal disordered structures,
both types of which are empirical laws of anomalous relaxation known so far.
Moreover the MC simulations of correlated random walks indicate that an
inertia of a particle does not have influence on the long-time behavior
of the anomalous relaxation.
-
J. Non-Cryst. Solids 198-200, 507-511 (1996).
-
-
Complexity in plasma: From self-organization to geodynamo
-
T. Sato, S. Bazdenkov, B. Dasgupta, S. Fujiwara, A. Kageyama,
S. Kida T. Hayashi, R. Horiuchi, H. Miura, H. Takamaru, Y. Todo,
K. Watanabe and T.-H. Watanabe
-
A central theme of ``Complexity'' is the question of the creation of
ordered structure in nature (self-organization). The assertion is
made that self-organization is governed by three key processes, i.e.,
energy pumping, entropy expulsion and nonlinearity. Extensive efforts
have been done to confirm this assertion through computer simulations
of plasmas. A system exhibits markedly different features in
self-organization, depending on whether the energy pumping is
instantaneous or continuous, or whether the produced entropy is
expulsed or reserved. The nonlinearity acts to bring a nonequilibrium
state into a bifurcation, thus resulting in a new structure along with
an anomalous entropy production. As a practical application of our
grand view of self-organization a preferential generation of a dipole
magnetic field is successfully demonstrated.
-
Phys. Plasmas 3(5), 2135-2142 (1996).
-
-
Anomalous relaxation in fractal and disordered structures
-
Susumu Fujiwara and Fumiko Yonezawa
-
For the purpose of clarifying the mechanisms of
the anomalous relaxation,
we carry out the Monte Carlo simulations of random walks in fractal
and disordered structures.
From our calculations of the relaxation functions and the complex
susceptibilities, we find that
the anomalous relaxation is of the Cole-Cole type in fractal
structures while it is of the stretched-exponential type
in non-fractal disordered structures.
A most interesting aspect indicated by the results of our work is that
the concept of fractal, originally introduced from purely
mathematical point of view, is shown to play important roles in
understanding some properties of realistic physical systems.
-
Int. J. Mod. Phys. B 10(26), 3561-3568 (1996).
-
-
Molecular Dynamics Simulations of Anomalous Relaxation in a Binary
Lennard-Jones System
-
Susumu Fujiwara and Fumiko Yonezawa
-
The anomalous relaxation in normal and supercooled liquids
is studied by molecular dynamics (MD) simulations of
a simple Lennard-Jones binary mixture
for various temperatures and wave numbers.
Our MD simulations show that the anomalous structural relaxation
of a stretched-exponential type appears not only in a supercooled
liquid region but also in a normal liquid region.
This fact indicates that the anomalous relaxation is not a feature
characteristic of supercooled liquids alone,
but rather it is a phenomenon to be found in broader
categories of disordered systems.
-
Phys. Rev. E 54(1), 644-649 (1996).
-
-
Monte Carlo Simulations of Anomalous Relaxation in Percolating Systems
-
Susumu Fujiwara and Fumiko Yonezawa
-
By Monte Carlo simulations of random walks in percolating systems, we show
that the anomalous relaxation observed in disordered systems can be ascribed
to the restricted geometry allowed for diffusion. In the fractal region
corresponding to the concentration p=pc, the
relaxation function has scaling properties and becomes the Cole-Cole type,
while, in the nonfractal disordered region corresponding to
p>pc, the relaxation function becomes the
stretched-exponential and Cole-Cole type, respectively, when
 and
and  .
From detailed analyses, we give a microscopic explanation for these two
types of relaxation functions known so far only as the empirical laws.
.
From detailed analyses, we give a microscopic explanation for these two
types of relaxation functions known so far only as the empirical laws.
-
Phys. Rev. Lett. 74(21), 4229-4232 (1995).
-
-
Anomalous Relaxation in Fractal Structures
-
Susumu Fujiwara and Fumiko Yonezawa
-
For the purpose of studying some interesting properties of anomalous
relaxation in fractal structures, we carry out Monte Carlo simulations of
random walks on two-dimensional fractal structures (Sierpinski carpets
with different cutouts and site-percolation clusters in a square lattice
at the critical concentration). We find that the relaxation is of the
Cole-Cole type [J. Chem. Phys. 9, 341 (1941)] which is one of the
empirical laws of anomalous relaxation. Scaling properties are found in
the relaxation function as well as in the particle density. We also find
that, in structures with almost the same fractal dimension, relaxation
in structures with dead ends is slower than that in structures without them.
This paper ascertains that the essential aspects of the anomalous relaxation
due to many-body effects can be explained in the framework of the one-body
model.
-
Phys. Rev. E 51(3), 2277-2285 (1995).
-
-
Slow dynamics in supercooled liquids: molecular dynamics simulations
-
Fumiko Yonezawa and Susumu Fujiwara
-
The relaxation processes in supercooled liquids were studied by molecular
dynamics simulations of simple systems such as a simple Lennard-Jones binary
mixture. Our simulations show (1) the existence of three stages of relaxation
(microscopic, β and
α-relaxation), (2) the scaling behavior of the
susceptibility at the minima in the intermediate frequency region and the
α-relaxation peak in the low frequency side,
and (3) that the mechanisms
of α- and
β-relaxation
are related to each other and independent of
temperature. Although these features have only been observed experimentally
so far in complex fluids, our results indicate that simple systems reveal the
essential features of anomalous relaxation.
-
Mat. Sci. & Eng. A 178, 23-27 (1994).
-
-
Monte Carlo simulations of anomalous relaxation in a-Si
---random walk in spaces of fractal dimension ---
-
Susumu Fujiwara, Sohei Gomi, Kazuo Morigaki and Fumiko Yonezawa
-
By carrying out Monte Carlo simulations of random walks in structures of
fractal dimension, we show that the anomalous relaxation observed in
amorphous systems including a-Si is ascribable to the restricted space allowed
for diffusion in disordered systems. We calculate the mean square displacement
as well as the exponent and relaxation time of the stretched-exponential
decay in the Fourier components of the density. Discussion is given about the
relations between the time dependences of these quantities and the fractal
dimension.
-
J. Non-Cryst. Solids 164-166, 301-304 (1993).
-
-
Stochastic transport model for diffusive properties in amorphous systems
-
Sohei Gomi, Susumu Fujiwara and Fumiko Yonezawa
-
The diffusive properties in amorphous systems are studied by Monte Carlo
simulations of a continuous time random walk on a one dimensional lattice.
When the probability distribution for the time interval between jump events
satisfies the power law
t−1−α,
the relaxation becomes anomalous of a stretched-exponential type when
α<1,
while transport phenomena similar to normal diffusion are observed when
α>1.
Scaling properties both in real and reciprocal space are ascertained.
-
J. Non-Cryst. Solids 164-166, 465-468 (1993).
-
Last modified on 06/25/26
Copyright (C) 1996-2026
Susumu Fujiwara